Journal list menu

 Issue
IssueAngewandte Chemie: Volume 102, Issue 12
i, fmi-A426, 1415-1544Dezember 1990
Export Citations
Download PDFs
Titelbild
Impressum
Graphisches Inhaltsverzeichnis
Aufsätze
Knüpfung von CC-Bindungen durch Addition von Carbenium-Ionen an Alkene: Kinetik und Mechanismus†
- Pages: 1415-1428
- First Published: Dezember 1990
Ein Schlüsselschritt zahlreicher Synthesen in der Organischen und Makromolekularen Chemie, die Addition von Carbenium-Ionen an CC-Doppelbindungen, wird am Beispiel der Lewis-Säure-induzierten Umsetzungen von Alkylchloriden mit Alkenen mechanistisch analysiert. Stereochemische und kinetische Untersuchungen deuten einen wenig verbrückten, Produkt-ähnlichen Übergangszustand an. Reaktivitäts-Selektivitäts-Beziehungen über einen Reaktivitätsbereich von acht Zehnerpotenzen zeigen, daß die Struktur des Übergangszustands nur durch Substituentenvariation in unmittelbarer Nähe des Reaktionszentrums verändert wird.
Zuschriften
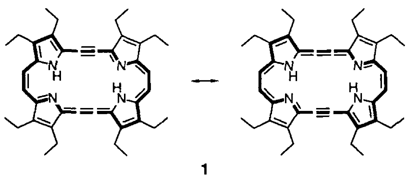
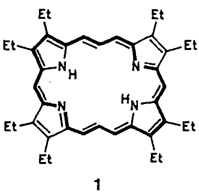
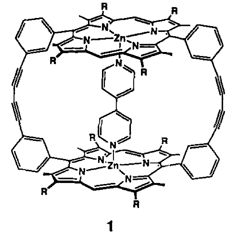
Acetylen-Cumulen-Porphyrinoide†‡
- Pages: 1429-1431
- First Published: Dezember 1990
Die Kombination von Porphyrin- und Acetylen-Chemie führt zu interessanten neuen Molekülen. Dies lehrt die Synthese des ersten Acetylen-Cumulen-Porphyrinoids 1, das als gestrecktes Porphycen betrachtet werden kann.
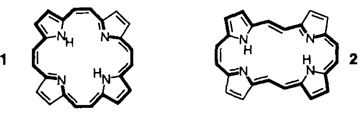
Die Porphyrin-Homologen: [22]Porphyrin (2.2.2.2), ein „gestrecktes Porphycen”︁†
- Pages: 1431-1434
- First Published: Dezember 1990
Die enge strukturelle Verflechtung von Porphyrin- und Porphycen-System wird mit dem [22]Porphyrin(2.2.2.2) 1/2 besonders augenfällig demonstriert: [22]Porphyrin(2.2.2.2) existiert aus Spannungsgründen nicht als das Isomer mit Porphyrin-Symmetrie (D2h) 1, sondern liegt als gestrecktes Porphycen 2 vor.
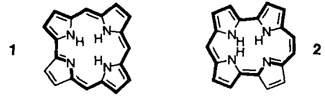
Isocorrole: Neuartige tetrapyrrolische Makrocyclen†‡
- Pages: 1434-1437
- First Published: Dezember 1990
Die Ringkontraktion eines Porphycenderivats ergibt ein Isocorrolderivat, dessen Ringgerüst deutlich stärker als das von Corrol von der Planarität abweicht. Nach spektroskopischen Befunden hat man es dennoch mit einer aromatischen Verbindung zu tun. 1 und 3 sind die Stammverbindungen Corrol bzw. Isocorrol.
Synthese eines bisvinylogen Octaethylporphyrins†‡
- Pages: 1437-1439
- First Published: Dezember 1990
Hans Fischers Octaethylporphyrin (OEP) ist bis heute von allen synthetischen Porphyrinen das bestverfügbare und meistuntersuchte. Durch eine stufenarme Synthese mit hohen Ausbeuten konnte nun das bisvinyloge OEP 1 gewonnen werden. Dieses ist durch chemische Stabilität, gute Zugänglichkeit und die bisher bei einem organischen Farbstoff intensivste UV/VIS-Absorption ein System mit vielseitigen Perspektiven.
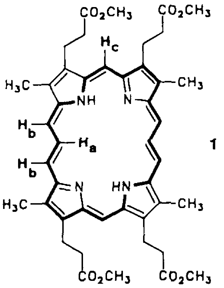
[22]Corproporphyrin II für die photodynamische Therapie†‡
- Pages: 1439-1441
- First Published: Dezember 1990
Hämatoporphyrin hat Bedeutung für eine photodynamische Tumortherapie (PDT). Funktionelle Nachteile der „oberen”︁ Molekülhälfte dieses Photosensibilisators, wie chemische Instabilität und Stereoisomerie, stehen der medizinischen Anwendung jedoch noch im Wege. Das neue, aus zwei „unteren”︁ Molekülhälften aufgebaute [22]Coproporphyrin II 1 ist durch Stabilität, sterische Einheitlichkeit und längerwellige, intensive UV/VIS-Banden charakterisiert. 1 erwies sich als aromatisches [22]Annulen.
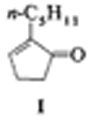
2-Pentylcyclopent-2-en-1-on durch katalytische Pauson-Khand-Reaktion†
- Pages: 1441-1443
- First Published: Dezember 1990
Nur 0.0022 Moläquivalente des Katalysators [Co2(CO)8] werden benötigt, um mit einer Wechselzahl von etwa 220 das Cyclopentenon 1 aus 1-Heptin, Ethylen und CO herzustellen. Diese erste katalytische Pauson-Khand-Reaktion (PKR) mit einem nichtgespannten Alken liefert 1, eine Vorstufe des für die Parfümindustrie wichtigen Methyl-trans-dihydrojasmonats, in etwa 50% Ausbeute. Auch die wichtigsten Nebenprodukte einer PKR wurden hier identifiziert und mögliche Wege ihrer Bildung vorgeschlagen.
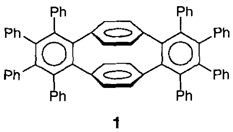
Ein Syntheseäquivalent für [2.2]Paracyclophan-1,9-diin: Octaphenyl-1:2,9:10-dibenzo[2.2]paracyclophan-1,9-dien und seine Reduktion zum Hexaanion†‡
- Pages: 1443-1445
- First Published: Dezember 1990
Das leicht zugängliche Tetrabrom[2.2]paracyclophan läßt sich mit Kalium-tert-butylalkoholat in Tetrahydrofuran schrittweise vierfach dehydrobromieren. Die dabei gebildeten [2.2]Paracyclophanine werden von Furan, Diphenylisobenzofuran und Tetraphenylcyclopentadienon zu entsprechenden Cycloaddukten effizient abgefangen. Mit besonders hoher Ausbeute (69%) wird das Octaphenydibenzo[2.2]paracyclophan-1,9-dien 1 isoliert, das formal aus zwei Hexaphenylbenzol-Einheiten besteht.
Aminosäureester als chirale Hilfsgruppen in Lewis-Säure-katalysierten Umsetzungen elektronenreicher Siloxydiene mit Iminen†
- Pages: 1445-1447
- First Published: Dezember 1990
Tandem-Mannich-Michael-Reaktionen und Aza-Diels-Alder-Reaktionen gehen die Aminosäureester-Imine 1 in Gegenwart unterschiedlicher Lewis-Säuren mit dem Danishefsky-Dien 2 bzw. dem Brassard-Dien 3 ein. Die Produkte entstehen mit guten Ausbeuten und hohen Diastereomerenüberschüssen. Durch Abspaltung der chiralen Hilfsgruppen werden daraus ungesättigte Lactame wie 4 und Enaminone wie 5 erhalten, die als Synthesebausteine Verwendung finden.

Darstellung und transmissionselektronen-mikroskopische Untersuchung von Bi(Pb)-Sr-Ca-Cu-O-Supraleitern†
- Pages: 1448-1450
- First Published: Dezember 1990
Durch einen hohen Anteil an Baufehlern, z. B. durch wechselnde Schichtabfolgen, sind die Realstrukturen der (BiO)2Sr2Can-1CunO2n+2-Supraleiter gekennzeichnet, wie hochaufgelöste transmissionselektronenmikroskopische Aufnahmen zeigen. Art und Umfang der Defekte können sich bei den einzelnen Ansätzen erheblich unterscheiden, so daß die Schwierigkeit, reproduzierbare Probeneigenschaften zu erhalten, verständlich wird.
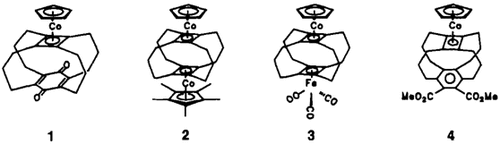
Aufbau metallstabilisierter, vierfach überbrückter Cyclobutadienophane†
- Pages: 1450-1452
- First Published: Dezember 1990
Ausgehend von 5-Cyclodecinol lassen sich nach einem Baukastenprinzip metallstabilisierte, vierfach überbrückte Cyclobutadienophane wie 1–4 aufbauen.
Doppelt überbrückte Prisman-, Dewar-Benzol- und Benzol-Derivate aus Cyclooctin und 1,8-Cyclotetradecadiin: en route zu Propella[n3]prismanen†
- Pages: 1452-1454
- First Published: Dezember 1990
Das hochgespannte Dewar-Benzol 1 sowie das Prisman 2 lassen sich in wenigen Stufen aus Cyclooctin erhalten. 2 entsteht durch Bestrahlung von 1 mit langwelligem Licht; bei λ = 250 nm entstehen nur Polymere, vermutlich über das entsprechende [5]Paracyclophan.
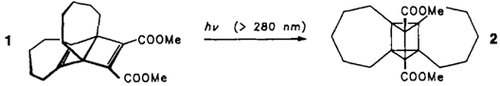
[Cu4(ddtp)2Br4], ein ungewöhnlicher, vierkerniger CuI-Komplex mit verbrückenden Thioether- und Bromidliganden†
- Pages: 1454-1455
- First Published: Dezember 1990
Eine spontane Reduktion von CuII zu CuI findet statt, wenn CuBr2 mit dem dreizähnigen SN2-Liganden ddtp in siedendem Ethanol/Aceton umgesetzt wird. Der entstehende Cu4-Titelkomplex enthält Thioetherbrücken mit großen Cu-S-Abständen und Cu-S-Cu-Winkeln (160.6°!). Die Cu2Cu2-Rechteckstruktur ist für CuI-Komplexe präzedenzlos. [Cu4(ddtp)2Br4] kann als Referenzverbindung für EXAFS-Untersuchungen an blauen Kupferproteinen dienen.
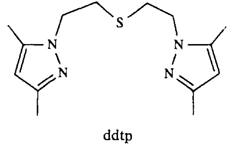
Erzeugung eines konfigurationsstabilen, chiralen Benzyllithium-Derivates und kapriziöse Stereochemie seiner elektrophilen Substitution†‡
- Pages: 1455-1456
- First Published: Dezember 1990
Je nach Elektrophil unter Retention oder Inversion der Konfiguration erfolgt die Substitution der in Lösung konfigurationsstabilen chiralen Benzyllithium-Verbindung 2, die durch Deprotonierung des entsprechenden optisch aktiven Benzylcarbamates 1 generiert wurde. Die Produkte 3 sind als Synthesebausteine von Interesse. Cb = C (=O)NiPr2.
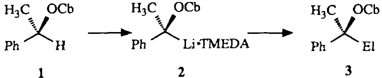
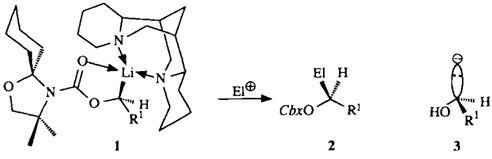
Chirale Lithium-1-oxyalkanide durch asymmetrische Deprotonierung; enantioselektive Synthese von 2-Hydroxyalkansäuren und sekundären Alkanolen†
- Pages: 1457-1459
- First Published: Dezember 1990
Eine allgemeine, sehr einfache präparative Lösung für chirale Synthone des Typs 3 bieten die Lithiumkomplexe 1. Sie werden durch asymmetrische Deprotonierung der entsprechenden prochiralen „nichtaktivierten”︁ Alkylcarbamate mit sec-Butyllithium/(-)-Spartein erzeugt. 1 läßt sich durch Elektrophile stereospezifisch zu den geschützten Hydroxy-Derivaten 2 (> 95 ee) substituieren, und die Cbx-Gruppe ist leicht abspaltbar. R1 = Alkyl, Cbx = Carbamoylrest von 1.
Isotopeneffekte bei der α-Olefineinschiebung in Zirconocen-Polymerisationskatalysatoren: Hinweise auf einen Übergangszustand mit α-agostischer Wechselwirkung†‡
- Pages: 1459-1460
- First Published: Dezember 1990
Die Hydrodimerisierung von trans- und von cis-1-Deuterio-1-hexen mit dem chiralen Präkatalysator rac-[Ethylenbis(tetrahydroindenyl)]zirconiumdichlorid zu den threo- und erythro-Isomeren von 6-Deuterio-5-deuteriomethylundecan ergab Diastereomerenverhältnisse von ca. 2.4:1, die sich je nach (E)- oder (Z)-Konfiguration des Olefins umkehren. Die Befunde bestätigen Beobachtungen von Pino et al., daß α-Olefine sich in Zr-H- und Zr-Alkyl-Bindungen mit entgegengesetzter enantiofacialer Orientierung einschieben.
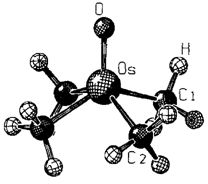
Organoosmiumoxide — gezielte Synthesen und Strukturen†
- Pages: 1460-1463
- First Published: Dezember 1990
Nicht nur das bisher einfachste Alkylosmiumoxid, Me4OsO, das in der Gasphase stabil ist, und dessen Struktur (Bild rechts) durch Elektronenbeugungsanalyse aufgeklärt werden konnte, sondern auch das thermolabile Et4OsO und ein gemischt alkyliertes Derivat ließen sich in guten Ausbeuten herstellen. Ferner wird über das strukturell neuartige Trimer [{(py)OsO2Me2}3] mit planarem Os3O3-Sechsring berichtet.
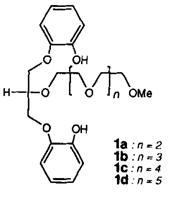
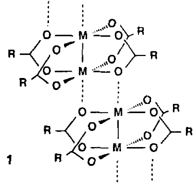
Neuartige Metalleinschlußverbindungen†
- Pages: 1463-1465
- First Published: Dezember 1990
Alle zehn Sauerstoffatome des acyclischen Oligoethers 1c beteiligen sich an der Koordination von Ba2⊕ im „Skorpion”︁-Komplex 1c · Ba(SCN)2 · Me2CO. Die elfte Koordinationsstelle besetzt das Acetonmolekül. Ein weiteres Kennzeichen des Komplexes sind starke O-H ⃛N-Wasserstoffbrücken zu den Thiocyanat-Gegenionen. Die Liganden 1 zeigen zudem eine Größenselektivität für Kationen mit Ionenradien von 1.2–1.3 Å, zu deren Erklärung Molecular-Modeling-Studien durchgeführt wurden.
Fe⊕-katalysierte Gasphasenoxidation von Ethan durch N2O†‡
- Pages: 1466-1468
- First Published: Dezember 1990
Die Übertragung des N2O-Sauerstoffatoms auf ein Fe(C2H4)⊕-Ion unter Bildung des Metallacyclus 1 ist der Schlüsselschritt der Titelreaktion. Dies folgt aus Ionen-Cyclotron-Resonanzexperimenten ebenso wie aus mechanistischen und thermochemischen Überlegungen. Nach Zerfall des „heißen”︁ Intermediats 1 zu Fe⊕ und Acetaldehyd [G1. (a)] kann ein neuer Katalysecyclus gestartet werden.
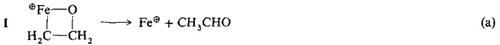
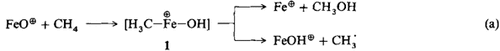
FeO⊕ aktiviert Methan†‡
- Pages: 1468-1469
- First Published: Dezember 1990
— wobei im wesentlichen FeOH⊕ + CH+3 sowie Fe⊕ + CH3OH entstehen. Zwar läuft diese Reaktion [G1. (a)] viermal langsamer als die in der vorstehenden Zuschrift beschriebene ab, dennoch stehen die Chancen für eine katalytische Aktivierung nicht schlecht, da das im Zuge der reduktiven Eliminierung aus der Zwischenstufe 1 regenerierte Fe⊕ durch N2O leicht wieder der Reaktion zugeführt werden kann.
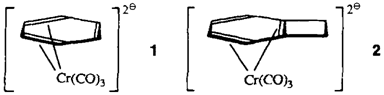
Darstellung und direkte NMR-spektroskopische Beobachtung von [(η4-aren)Cr(CO)3]-Dianionen†
- Pages: 1469-1471
- First Published: Dezember 1990
Lithiumsand statt Naphthalinkalium liefert bei der Synthese der Dianionen 1 und 2 aus den η6-koordinierten Neutralkomplexen reine Produkte, da keine Möglichkeit zu Austauschreaktionen mit Naphthalin besteht. Auffällig an 2 ist, daß die gespannteste Doppelbindung der Cyclobutabenzol-Einheit auch nach der Reduktion an das Metallzentrum gebunden bleibt.
GeII und SnII-Komplexe des [2.2.2]Paracyclophans mit dreifacher interner η6-Koordination†
- Pages: 1471-1473
- First Published: Dezember 1990
Deutlich fester als GeII ist SnII im Hohlraum des [2.2.2]Paracyclophans(L) gebunden, wie die Strukturanalysen der komplexen Kationen [LSn(AlCl4)]⊕ und [LGeCl]⊖ 1 zeigen. Während im Zinnkomplex das AlCl⊕4-Gegenion nur locker über ein Cl-Atom am Metallzentrum koordiniert ist (Sn-Cl 3.073 Å) und damit die erwartete trigonal-planare Koordination nur unwesentlich stört, führt die starke Ge-Cl-Bindung (Ge-Cl 2.224 Å) in 1 zu einer Abschwächung der Ge-Aren-Wechselwirkung und einer eher tetraedrischen Umgebung des Metallzentrums.

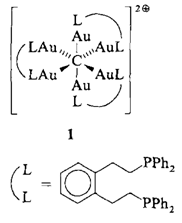
Aufbau des [CAu6]2⊕-Clusters mit einem maßgeschneiderten, die Oktaederkanten überspannenden Diphosphan†
- Pages: 1473-1475
- First Published: Dezember 1990
Über Chelatliganden verbrückte Digoldkomplexe eignen sich ebenfalls zur „molekularen Vergoldung”︁ von Kohlenstoffatomen, wie die Struktur des CAu6-Komplexkations 1 zeigt. Aufgrund der Asymmetrie der Ligandensphäre sind die Kationen chiral. Die Bildung von 1 demonstriert, daß das Aurophilie-Konzept auch auf zweizähnige Liganden erweitert werden kann.
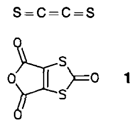
C2S2 (Ethen-1,2-dithion), ein Beispiel für die Verletzung der Hundschen Regel?†‡
- Pages: 1475-1477
- First Published: Dezember 1990
Die erste Verletzung der Hundschen Regel in einer Gleichgewichtsstruktur legen quantenchemische Rechnungen für C2S2 nahe, denn sie liefern entgegen der Erwartung einen Singulett-Grundzustand. Die experimentellen Befunde der IR- und UV-spektroskopischen Untersuchung des in einer Ar-Matrix aus Vorläuferverbindungen wie 1 erhaltenen C2S2 stimmen mit den Rechnungen gut überein.
Synthese cyclischer Porphyrin-Oligomere mit Aminen als Templaten†
- Pages: 1478-1480
- First Published: Dezember 1990
In 70% Ausbeute läßt sich das Dimer 1 durch Glaser-Kupplung herstellen, wenn die Reaktion in Gegenwart von 4,4′-Bipyridin als Templat durchgeführt wird. Mit einem Tripyridyltriazin als Templat entsteht bevorzugt das entsprechende Trimer (52%). Ohne Templat verläuft die Reaktion unselektiv: Dimer und Trimer entstehen nebeneinander in 20–25 bzw. 30–35% Ausbeute. Die Ausbeuten korrespondieren gut mit den Bindungseigenschaften dieser Käfigmoleküle, die als Vorstufen für Enzymmodelle von Interesse sind.
Lewis-Basen-gesteuerte Strukturvarianten in der Natriumamid-Chemie: Röntgenstrukturanalysen von Phenyl(2-pyridyl)natriumamid-Addukten mit Hexamethylphosphorsäuretriamid und mit Pentamethyldiethylentriamin†
- Pages: 1480-1481
- First Published: Dezember 1990
Sehr unterschiedliche Strukturen haben die dinuclearen Natriumamid-Addukte 1 und 2: In 1 sind die zweizähnigen Amid-Ionen terminal an die Na⊕-Ionen gebunden, in 2 verbrücken sie diese. Darüber hinaus ist bemerkenswert, daß HMPA in 1 verbrückend wirkt und daß in 2 das dreizähnige PMDETA nur mit zwei N-Atomen an Na⊕ koordiniert ist.

Decamere und dodecamere Natriumamide sowie ein Organonatriumamid mit Stapelstrukturen: [Na10(NMe2)10(tmeda)4], [Na12(NMe2)12(tmeda)4] und [Na12(NMe2)10(CH2C6H4Me)2(tmeda)4]†‡
- Pages: 1481-1484
- First Published: Dezember 1990
Ringstapelstrukturen weisen die oligomeren Natriumamide 1–3 auf, die bei Versuchen zur Metallierung von Kohlenwasserstoffen durch n-Butylnatrium in Gegenwart von Tetramethylethylendiamin entstanden. Entsprechend gebaute Lithiumamide sind nicht bekannt; vielmehr sollten hier Leiterstrukturen bevorzugt sein.
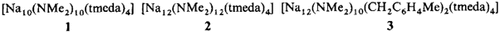
Synthese und Struktur von [Et2OZn(SC6H2tBu3)2], dem ersten T-förmigen Zink-Komplex†
- Pages: 1484-1485
- First Published: Dezember 1990
Die Stapelung von Arylgruppen scheint einmal mehr strukturbestimmend zu sein. Der Titelkomplex, der durch Umsetzung von [Zn(CH2SiMe3)2] mit zwei Äquivalenten 2,4,6-tBuC6H2SH in Gegenwart von Et2O zugänglich ist, hat einen S-Zn-S-Winkel von ca. 160° und Zn-S-Abstände von nur 2.196 Å. Der Zn-O-Abstand ist hingegen relativ groß und erklärt die leichte Abspaltbarkeit der Ether-Liganden. Zinkthiolate sind als Modellkomplexe für zinkhaltige Proteine von Interesse.
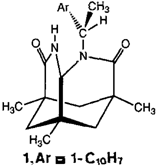
Asymmetrische Protonierung von Enolaten†
- Pages: 1485-1486
- First Published: Dezember 1990
Effiziente chirale Protonenquellen sind schon lange ein Wunschtraum der Organiker. Mit dem auf der Kempschen Tricarbonsäure basierenden Lactam 1 konnten jetzt cyclische Enolate mit einem Enantiomerenüberschuß (ee) von bis zu 91% protoniert werden. Ursache der hohen ee-Werte ist die asymmetrische Mikroumgebung der sauren NH-Gruppe.
Spontane Bildung stabiler Phosphino(silyl)carbene aus instabilen Diazoverbindungen
- Pages: 1486-1488
- First Published: Dezember 1990
Mehrere Wochen in Lösung stabil sind die carbenähnlichen Verbindungen 1, die aus Phosphanyldiazomethanen bereits bei T ≤ 35°C nahezu quantitativ entstehen, wenn der Tetramethylpiperidin(tmp)-Rest einer der Substituenten am Phosphor ist. Die Verbindungen 1 reagieren mit Ausnahme von 1a mit tBuNC zu den Keteniminen 2. a, R = NiPr2, R′ = Me; b, R = NMe2, R′ = Me; c, R = NMe2, R′ = iPr; d, R = Ph, R′ = Me.
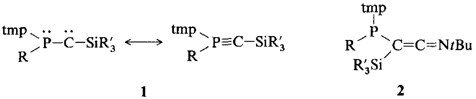
Oxidativer Abbau aromatischer Schadstoffe durch chemische Ligninase-Modelle auf Porphyrin-Basis†
- Pages: 1488-1490
- First Published: Dezember 1990
Mikrobiell nicht oder nur schwer abbaubare Chlorphenole und methoxylierte Arene werden in wenigen Minuten von einem Peroxidase-Modellsystem zu Chinonen oxidiert. Dieses Modellsystem wird durch Umsetzung sulfonierter Metalloporphyrin-Komplexe mit KHSO5 erhalten. Es oxidiert auch noch Arene wie 1,3,5-Trimethoxybenzol, das weder von Ligninase noch von Meerrettich-Peroxidase angegriffen wird.
Mehrfachbindungen zwischen Metallatomen in geordneten Aggregaten: Flüssigkristalle mit Mo-Mo-Vierfachbindungssystemen
- Pages: 1490-1491
- First Published: Dezember 1990
Der Temperaturbereich der Mesophasen der Carboxylatokomplexe [Mo2(O2CCnH2n+1)4] 1 hängt deutlich von der Länge der Kohlenwasserstoffkette ab. So geht 1, n = 5 (Hexanoat), bei 108°C in die Mesophase und erst bei 172°C in die isotrope Flüssigkeit über. Bei 1, n = 9 (Decanoat), betragen diese Werte 105 bzw. 116°C und bei 1, n = 11 (Dodecanoat), tritt keine Mesophase mehr auf.
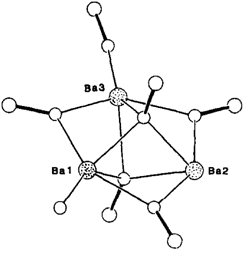
[Ba3(OSiPh3)6(THF)] · 0.5 THF — Synthese und Struktur eines dreikernigen Bariumsiloxids mit niedrig koordiniertem Barium
- Pages: 1492-1493
- First Published: Dezember 1990
Ein Dreieck aus Ba-Ionen mit zwei überdachenden μ3- und drei kantenverbrückenden μ2-OSiPh3-Liganden liegt in der zentralen Struktureinheit (Bild rechts) der Titelverbindung vor. Alle drei Ba-Ionen sind jedoch unterschiedlich koordiniert: So sorgt an Ba3 ein terminaler Siloxido-Ligand für Ladungsausgleich, während ein THF-Ligand an Ba1 gebunden ist. Der Dreikernkomplex bildet sich in 77% Ausbeute beim Einleiten von NH3 in eine Reaktionsmischung aus Ba-Granalien und Ph3SiOH in THF.
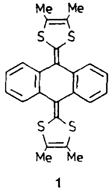
Röntgenstrukturanalyse sowie elektrische und magnetische Eigenschaften eines hoch leitfähigen 4: 1-Komplexes aus Tetracyanchinodimethan und einem Tetrathiafulvalen-Homologen†
- Pages: 1493-1495
- First Published: Dezember 1990
Trotz starker Verdrillung des konjugierten Systems reagiert das Tetrathiafulvalen-Homologe 1 mit dem Elektronenacceptor Tetracyanchinodimethan zum Dikation 12⊕. In 12⊕[(TCNQ)4]2⊕ ist das Anthracengerüst eingeebnet, und die Dithiolringe sind 86° gegen die Anthracenebene verdrillt. Die 1:4-Stöchiometrie des hoch leitfähigen Salzes (σ300K = 60 S cm−1) ist für TCNQ-Komplexe sehr ungewöhnlich.
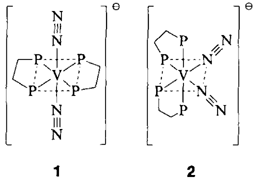
Vanadium(-I)-Stickstoffkomplexe mit end-on koordiniertem N2: funktionelle Modelle für die „alternative Nitrogenase”︁†
- Pages: 1495-1497
- First Published: Dezember 1990
Eine empfindliche Sonde für die Bildung der anionischen N2-Komplexe 1 und 2 bei der Reaktion von [VCl3(thf)3] mit N2, dmpe und Na ist ihr Zentralmetall. So ist das 51V-NMR-Signal des trans-Komplexes 1 ein Quintett bei δ = −1123. Wird die Reduktion unter 15N2 durchgeführt, so spalten die einzelnen Multiplett-Komponenten zusätzlich zu Tripletts auf. Damit liegt ein direkter Nachweis für den Einbau von Stickstoff in den Komplex vor. dmpe = Dimethylphosphinoethan.
Künstliche Rezeptoren für Kohlenhydratderivate†
- Pages: 1497-1499
- First Published: Dezember 1990
Deutliche Hinweise auf den Einschluß von Pyranosiden in den Makrocyclus 1 (R = H, R = Bn) lieferten NMR- und IR-spektroskopische Untersuchungen der Umsetzung mit 2 in CDCl3 sowie Kraftfeldrechnungen für das Methylanalogon von 2. Bei 1 handelt es sich somit um einen der ersten künstlichen Kohlenhydrat-Rezeptoren. Sein Hohlraum ist groß genug, um das Gastmolekül von allen Seiten durch gerichtete H-Brücken zu fixieren.
Lösliche, oligomere verbrückte Phthalocyaninatoeisen(II)-Komplexe
- Pages: 1499-1501
- First Published: Dezember 1990
Durchschnittlich 110 verknüpfte Metallomakrocyclen liegen in einem Oligomerstapel vor, wenn der Phthalocyaninatoeisen-Komplex 1, R1 = R2 = 2-Et-C6H12, mit Benzoldiisocyanid umgesetzt wird. Dieses und andere Oligomere konnten durch 57Fe-Mößbauer- und NMR-Spektroskopie charakterisiert werden.

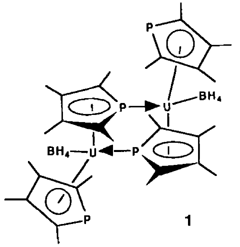
Synthese von Phospholyl(tetrahydroborato)uran-Komplexen. Kristallstruktur von [(η5-C4Me4P)2U(BH4)2]
- Pages: 1501-1502
- First Published: Dezember 1990
31P-NMR-chemische Verschiebungen von δ = 3471 (!) und δ = 727 spiegeln die ungewöhnliche Bindungssituation im dimeren, paramagnetischen Organouran(III)-Komplex 1 wider. Das extrem tieffeldverschobene Signal kann dem P-Atom des verbrückenden Phospholylliganden zugeordnet werden. 1 läßt sich mit TlBH4 zum Uran(IV)-Sandwich [(C4Me4P)2U(BH4)2] oxidieren, der bei Reduktion mit Na-Amalgam wieder 1 ergibt.
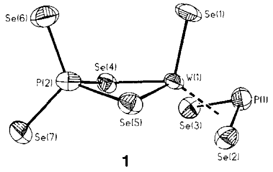
Synthese und Struktur von [Se=W(PSe4)(PSe2)]2⊖; ein Dianion mit einer heteroallylischen PSe2⊖-Einheit†
- Pages: 1502-1504
- First Published: Dezember 1990
Ein Nitrit-analoges, niederkoordiniertes Phosphorchalcogenid ist das überraschendste Strukturmerkmal des anionischen Wolframkomplexes 1. Die gewinkelte PSe⊖2-Einheit ist seitlich an das Wolfram gebunden, ein terminaler Seleno- und ein zweizähniger, tetraedrischer PSe3⊖4-Ligand vervollständigen die Koordinationssphäre des Metallzentrums.
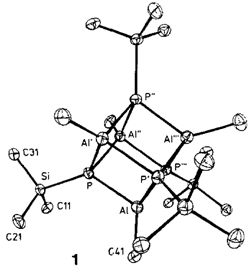
Ein Aluminaphosphacuban, ein neuer Vorläufer für Aluminiumphosphid†
- Pages: 1504-1505
- First Published: Dezember 1990
Nicht nur von ästhetischem Reiz ist der Al4P4-Würfel in [iBuAl(μ3-PSiPh3)]4 1. Sein Verhalten bei thermischer Belastung macht ihn auch als Vorläufer für den III/V-Halbleiter AlP interessant. Bereits bei 150°C werden die Substituenten als Ph3SiH und Isobuten abgespalten, Erhöhung der Temperatur auf 500°C führt zu reinem AlP.
s-trans-1,3-Diene als Liganden für Metall-Kationen der 8. Gruppe
- Pages: 1505-1507
- First Published: Dezember 1990
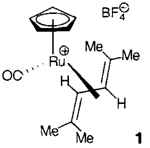
Bei der Koordination an Metallzentren nehmen 1,3-Diene sehr häufig eine s-cis-Konformation an. Der Ru-Komplex 1 ist der erste kationische s-trans-Dien-Komplex, der bei Raumtemperatur in Lösung stabil ist. Derartige Komplexe sind als reaktive Intermediate bei der katalytischen Umsetzung konjugierter Diene denkbar.
π/σ-Gleichgewichte in Metall-Komplexen organischer Carbonylverbindungen: Synthese und Struktur chiraler Rhenium-Komplexe [(η5-C5H5) Re (NO) (PPh3)(O=CHAr)]X†
- Pages: 1507-1509
- First Published: Dezember 1990
π/σ-Verhältnisse von > 96: < 4 bis 15:85 (bei 299 K) treten in den Komplexen 1 in Abhängigkeit vom Arylsubstituenten auf. Neben diesem starken elektronischen Einfluß konnten auch deutliche Lösungsmittel- und Temperatureinflüsse nachgewiesen werden. Diese Befunde sind für das Verständnis von Addukten aus Carbonylverbindungen und Metallkomplex-Fragmenten von großer Bedeutung.
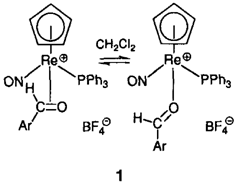
Thermodynamisch kontrollierte enantiofaciale Selektivität bei der Bindung von Aldehyden an chirale Metallkomplex-Fragmente – der Mechanismus der Interkonversion diastereomerer π-Aldehyd-Komplexe [(η5-C5H5) Re(NO)(PPh3)(η2-O = CHAr)]BF4†
- Pages: 1509-1511
- First Published: Dezember 1990
Je höher die π-Acidität des Aldehyd-Liganden desto ausgeprägter die chirale Erkennung bei seiner Bindung an das chirale Komplexfragment 1. Dies ist das Ergebnis Tieftemperatur-NMR-spektroskopischer Untersuchungen der Komplexe von 1 mit den aromatischen Aldehyden 2, die zudem zeigten, daß sich die diastereomeren π-Aldehyd-Komplexe aus 1 und 2 intramolekular über einen σ-Aldehyd-Komplex ineinander umwandeln.
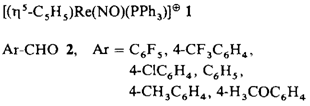
Neue chirale Tripod-Phosphane mit C3-Symmetrie†
- Pages: 1511-1513
- First Published: Dezember 1990
Gegenüber zweizähnigen chiralen Liganden mit C2-Symmetrie bewirken dreizähnige mit C3-Symmetrie in oktaedrischen Übergangsmetallkomplexen eine verminderte Anzahl nichtäquivalenter restlicher Koordinationsstellen. Dies läßt bei katalytischen Reaktionen eine erhöhte Enantioselektivität erwarten. Die Tripod-Liganden 1 und 2, die aus 2,5-Dimethyl-1-phenylphospholan leicht zugänglich sind, wurden als Liganden in Rh-Komplexen untersucht.
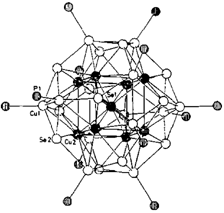
Neue Kupfercluster mit Se und PEt3 als Liganden: [Cu70Se35(PEt3)22] und [Cu20Se13(PEt3)12]†‡
- Pages: 1513-1516
- First Published: Dezember 1990
Der bisher größte röntgenkristallographisch charakterisierte Cluster, die Verbindung 1, entsteht zusammen mit wenig 2 (Struktur: Bild rechts) bei der Umsetzung von CuCl mit Se(SiMe3)2 und PEt3. Zwischenstufe auf dem Weg zu 1 könnte ein Cu9-Cluster sein. Beim Erhitzen von 1 auf über 160°C bildet sich Cu2Se.
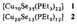
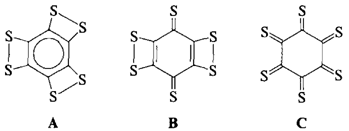
Die Struktur von cyclischem C6S6 und C6O6†‡
- Pages: 1516-1517
- First Published: Dezember 1990
Nicht aromatisch, sondern chinoid ist das energetisch günstigste Isomer des in der Gasphase nachgewiesenen C6S6. Die Energiedifferenz zwischen A und B wurde durch ab-initio-Rechnungen zu 10.9 kcal mol−1 bestimmt. Nahezu energiegleich mit B ist dagegen das Hexathioketon C, das in einer Sesselkonformation vorliegen sollte.
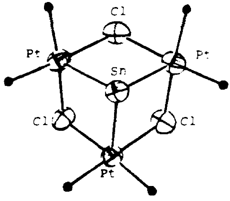
(NBu4) Sn{Pt(μ-Cl)(C6F5)2}3ein ungewöhnlicher Cluster mit drei PtII → SnII-Bindungen†
- Pages: 1518-1519
- First Published: Dezember 1990
Ein gewellter sechsgliedriger Pt3Cl3-Ring, der über seine basischen Pt-Zentren ein „nacktes”︁ SnII-Zentrum koordiniert, so läßt sich der Clusterkern (Bild rechts) der Titelverbindung beschreiben. Außer den Pt-Sn-Wechselwirkungen tragen noch kurze Kontakte zwischen SnII und den o-F-Atomen der C6F5-Liganden am Platin zur Stabilisierung dieses ungewöhnlichen Komplexanions bei.
CsF · Br2, eine Alkalimetallhalogenid-Intercalationsverbindung†
- Pages: 1519-1520
- First Published: Dezember 1990
Bei 80°C nimmt Caesiumfluorid Brom auf, und zwar im Verhältnis 1:1. Dabei weitet sich das Wirtsgitter parallel zu einer der (100)-Flächen auf (Bild rechts). Der Verlust an Gitterenergie wird bereits durch eine sehr schwache Br ⃛F⊖-Wechselwirkung in der Größe von wenigen kJ mol−1 kompensiert.
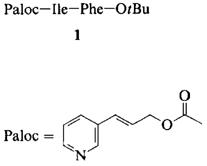
Der 3-(3-Pyridyl)allyloxycarbonyl(Paloc)-Rest — eine stabile, unter neutralen Bedingungen abspaltbare Aminoschutzgruppe für Peptidsynthesen in organischen Medien und in Wasser†
- Pages: 1520-1522
- First Published: Dezember 1990
Stabil selbst gegen starke Säuren ist die Palocgeschützte Aminofunktion in Dipeptidestern wie 1. Dies ermöglicht die selektive, quantitative Abspaltung der tBu-Gruppe mit HCl in Et2O/CH2Cl2. Umgekehrt gelingt die gezielte Ablösung der Paloc-Schutzgruppe durch Palladium(0)-katalysierte Allylübertragung auf schwach basische Nucleophile wie N-Methylanilin. Bei dieser Reaktion bleiben viele Schutzgruppen intakt, d. h. empfindliche Strukturen werden nicht zerstört.
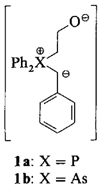
Neue Benzyl(β-oxido)phosphor- und -arsen-Ylide: Reaktivität und Stereoselektivität bei der Wittig-Reaktion†
- Pages: 1522-1523
- First Published: Dezember 1990
Wittig-Reaktionen mit semistabilisierten Yliden verlaufen meist stereounselektiv. Unter konsequenter Berücksichtigung bekannter Fakten wurden nun die Benzyl-Ylide 1a und 1b hergestellt und mit Aldehyden umgesetzt. Das Arsen-Ylid 1b lieferte bei der Reaktion mit Piperonal und Hexanal jeweils 100% des (E)-Olefins (GC-Analyse).
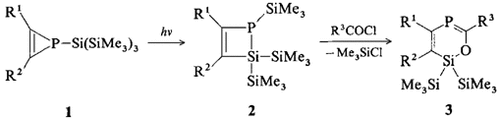
Eine 1-Silyl-1H-phosphiren/1,2-Dihydro-1,2-phosphasilet-Umlagerung — der entscheidende Schritt zur Synthese von Heterocyclen mit 2-Phospha-1,3-dien-Einheiten†‡
- Pages: 1523-1525
- First Published: Dezember 1990
Photochemisch lagert sich das Silyl-substituierte 1H-Phosphiren 1 zum kristallinen Dihydrophosphasilet 2 um. Wird dieses am Phosphoratom acyliert, so entsteht durch spontane 1,3-Silylverschiebung 3, ein Heterocyclus mit einer 2-Phospha-1-3-dien-Einheit. R1 = R3 = tBu, R2 = Ph.
Induktion und Variation von Chiralität in discotisch-flüssigkristallinen Polymeren
- Pages: 1525-1528
- First Published: Dezember 1990
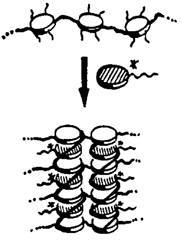
Wegen ihrer Ähnlichkeit zu helicalen Polymeren und für einen Vergleich mit chiralen stabförmigen Flüssigkristallen sind chirale discotisch-flüssigkristalline Polymere interessant. Sie wurden nun auf mehrere Arten zugänglich gemacht, z. B. durch Dotierung achiraler Donor-Polymere mit chiralen Acceptoren (siehe Bild). Die Chiralität der discotischen Phasen wurde an dünnen Filmen CD-spektroskopisch untersucht.
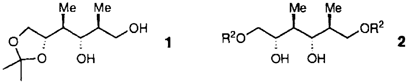
Enantio- und regiokontrollierte Synthese eines zentralen Ionophor-Antibiotica-Bausteines durch sequentielle Öffnung zweier Epoxidringe mit Cuprat-Reagentien†‡
- Pages: 1529-1530
- First Published: Dezember 1990
Die sequentielle Öffnung zweier vicinaler Epoxidgruppen durch Cuprat-Reagentien in einem Schritt ist die Schlüsselreaktion beim Aufbau der Stereotetraden 1 und 2. Als Edukt für beide Verbindungen, die in mehreren Ionophor-Antibiotica enthalten sind, dient D-Mannit.
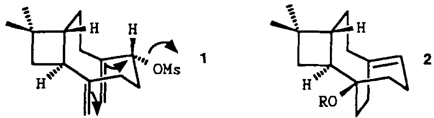
Eine stereoselektive π-Cyclisierung zu einem kombinierten Brückenkopfolefin/-alkohol†
- Pages: 1530-1532
- First Published: Dezember 1990
Brückenkopfolefin- und Brückenkopfalkohol-Einheit in einem Molekül entstehen gleichzeitig bei der Solvolyse des Mesylats 1 zu 2 (R = H). Die Synthese von 1 aus Caryophyllenepoxid gelang erstmals stereoselektiv und in guter Ausbeute. Eine Röntgenstrukturanalyse des 4-Brombenzoats von 2 bestätigt die Strukturzuordnung.
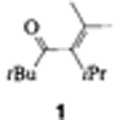
Die sterisch gehinderte Rotation um die C–C–Bindung eines acyclischen α,β-ungesättigten Ketons: 2,2,5-Trimethyl-4-isopropylhex-4-en-3-on†
- Pages: 1532-1533
- First Published: Dezember 1990
Eine Aktivierungsbarriere von 45 kJ mol−1 folgt aus den 1H-NMR-Spektren für die erste nachgewiesene gehinderte Rotation um die sp2-sp2-Einfachbindung eines acyclischen α,β-ungesättigten Ketons (1). Die Intensitäten der UV-Banden der π → π*- und n → π*-Übergänge sind mit einer verdrillten Konformation von 1 in Einklang. Molekülmechanikrechnungen lassen allerdings eine deutlich höhere Aktivierungsbarriere erwarten, was die schlechte Parametrisierung für sp2-sp2-Einfachbindungen belegt.
Neue Bücher
Chemiker als Literaten Cantor's Dilemma. Von C. Djerassi Doubleday, New York (NY, USA) 1989. 230 S., geb. $ 18.95. — ISBN 0–385–26183–7/für Europa: Macdonald & Company, London (UK) 1990. 7pound; 12.95. — ISBN 0–356–19050–1
- Pages: 1533-1534
- First Published: Dezember 1990
Gaps and Verges. Von R. Hoffmann. University of Central Florida Press, Orlando (FL, USA) 1990. 88 S., geb. $ 14.95. — ISBN 0–8130–0943-X
- Pages: 1534-1535
- First Published: Dezember 1990
pH-Messungen. Grundlagen, Methoden, Anwendungen, Geräte. Von H. Galster. VCH Verlagsgesellschaft, Weinheim 1990. XV, 322 S., geb. DM 184.00. — ISBN 3–527–27836–2
- Pages: 1535-1536
- First Published: Dezember 1990
Aquatische Chemie. Eine Einführung in die Chemie Wässriger Lösungen und in die Chemie natürlicher Gewässer. Von L. Sigg und W. Stumm. vdf, Verlag der Fachvereine an den Schweizer Hochschulen und Techniken, Zürich 1989. 388 S., Broschur SFr. 46.00.—ISBN 3–7281–1729–3
- Page: 1536
- First Published: Dezember 1990
Structure and Reactivity in Reverse Micelles. (Reihe: Studies in Physical and Theoretical Chemistry, Vol. 65). Von M. P. Pileni Herausgegeben Elsevier, Amsterdam 1989. XVIII, 379 S., geb. Hfl. 285.00. — ISBN 0–444–88166–2
- Pages: 1536-1537
- First Published: Dezember 1990
Chemically Modified Carbon Fibers and their Applications. Von I. N. Ermolenko I. P. Lyubliner N. V. Gulko VCH Verlagsgesellschaft, Weinheim/VCH Publishers, New York 1990. X, 304 S., geb. DM 184.00. — ISBN 3–527–26927–4/0–89573–873–2
- Pages: 1537-1538
- First Published: Dezember 1990
Lectins. Von N. Sharon und H. Lis Chapman & Hall, London 1989. VII, 127 S., geb. £ 19.50. — ISBN 0–412–27380–2
- Page: 1538
- First Published: Dezember 1990
Mycotoxins. Chemical, Biological, and Environmental Aspects. (Reihe: Bioactive Molecules Vol. 9). Von V. Betina Elsevier, Amsterdam 1989. 438 S., geb. HFl. 295.00. — ISBN 0–444–98885–8
- Pages: 1538-1539
- First Published: Dezember 1990
ABC Geschichte der Chemie. Herausgegeben von einem Autorenkollektiv, federführende Herausgeber: S. Engels und R. Stolz. VEB Deutscher Verlag für Grundstoffindustrie, Leipzig 1989. 496 S. + 15 S. Anhang, geb. (Leinen) DM 60.00.—ISBN 3–342–00118–6
- Page: 1539
- First Published: Dezember 1990
Sensors — A Comprehensive Survey. Reihenherausgeber: W. Göpel,VJ. Hesse und J. N. Zemel VCH Verlagsgesellschaft, Weinheim. Vol. 1: Fundamentals and General Aspects. Bandherausgeber: T. Grandke und W. H. Ko 1989. XXXIII, 641 S., geb. DM 350.00 (Subskriptionspreis: DM 290.00). — ISBN 3–527–26767.0; Vol. 5: Magnetic Sensors. Bandherausgeber: R. Boll K. J. Overshott XII, 513 S., geb. DM 350.00 (Subskriptionspreis: DM 290.00). — ISBN 3–527–26771–9
- Pages: 1539-1540
- First Published: Dezember 1990
Analytical NMR. Von L. D. Field und S. Sternhell Herausgegeben von J. Wiley, Chichester 1989. VIII, 250 S., geb. £ 32.95. — ISBN 0–471–91714–1
- Pages: 1540-1541
- First Published: Dezember 1990
Biomineralization, Chemical and Biochemical Perspectives. Herausgegeben S. Mann J. Webb und R. J. P. Williams VCH Verlagsgesellschaft, Weinheim/VCH Publishers, New York 1989. XIV, 541 S., geb. DM 274.00. — ISBN 3–527–26750–6/0–89573–672–1
- Page: 1541
- First Published: Dezember 1990
Molecular Structure of Organosilicon Compounds. (Reihe: Ellis Horwood Series in Organic Chemistry). Von E. Lukevics O. Pudova und R. Sturkovich Ellis Horwood, Chichester 1989. 359 S., geb. £ 69.95. — ISBN 0–7458–0692–9
- Pages: 1541-1542
- First Published: Dezember 1990
Kunststoffverbundsysteme — Grundlagen, Anwendung, Verarbeitung, Prüfung. Von R. Janda VCH Verlagsgesellschaft, Weinheim 1990, XII, 319 S., geb. DM 144.00. — ISBN 3–527–27864–8
- Page: 1542
- First Published: Dezember 1990
Schreiben und Publizieren in den Naturwissenschaften. Von H. F. Ebel und C. Bliefert VCH Verlagsgesellschaft, Weinheim 1990, XIV, 442 S., Broschur DM 48.00. — ISBN 3–527–27990–3
- Pages: 1542-1543
- First Published: Dezember 1990
Houben-Weyl. Methoden der Organischen Chemie. 4. Auflage, Erweiterungs- und Folgebände. C-Radikale, Teil 1 und 2. (Bände E 19 a). Herausgegeben Von M. Regitz und B. Giese Thieme, Stuttgart 1989. Teil 1: S. 1–716; Teil 2: S. 717–1567. Geb. DM 1960.00 (Subskriptionspreis: DM 1764.00). — ISBN 3–13–218904–9
- Pages: 1543-1544
- First Published: Dezember 1990
Sign up for email alerts
Tools
Eine Zeitschrift der
Gesellschaft Deutscher Chemiker
More from this journal
Classifieds: Jobs and Awards, Products and Services
- Angewandte Chemie
- 4 November 2024
Graphical Abstract

Are you looking for a (new) job? Do you know someone who deserves an award? Are you trying to find a product or service to make your work more efficient? Our partners can help.
Related Titles



 IssueVolume 97, Issue 7
IssueVolume 97, Issue 7Special Issue: Membrantechnik
July 2025Guest Editor(s):
Deutsche Gesellschaft für Membrantechnik