Journal list menu


The "Bürgenstock Conference" is held annually in Brunnen, overlooking Lake Lucerne. The main program, which is not announced until the event starts, consists of 13 hour‐long plenary lectures with 30 minutes for discussion, as well as short presentations and poster sessions. The 54th conference took place from May 5‐ 9, 2019 with President Véronique Gouverneur (University of Oxford), Vice‐President Janine Cossy (ESPCI-Paris), and Guest of Honor John Brown (University of Oxford). In this Virtual Issue, we feature a selection of recent papers by the speakers and authors related to the conference, and some pieces about the history and philosophy of the conference.
Export Citations
Download PDFs
Editorial
Half a Century of the Bürgenstock Conference: A Pilgrim’s Tale
- First Published: 02 April 2015

“…︁ The Bürgenstock program always allows for important scientific discussion. It is clear that this meeting is valuable for the scientific community as it is on one hand important for its deep historical significance, and on the other hand crucial for what it preserves of great scientific tradition for future generations. Every meeting contained a lesson learned and further brought together the family of stereochemists …︁” Read more in the Editorial by Jay S. Siegel.
Essay
50 Years in the Sun of Bürgenstock—On the Success Factors of a Famous Conference
- First Published: 20 March 2015

The secret of success: This year the famous “Bürgenstock Conference” will take place for the 50th time. This conference has become internationally one of the, if not the, most highly regarded conference in chemistry, chemical biology, and physical chemistry. What are the success factors of this conference? These as well as a number of perhaps more hidden figures and facts are discussed.
Meeting Reviews

Scientific Fireworks to Celebrate the 50th Anniversary of the Bürgenstock Conference†
- First Published: 26 June 2015

Thought-provoking for half a century: The famous Bürgenstock Conference recently celebrated its 50th anniversary, which was marked with outstanding science and vibrant discussions in a distinctively stimulating setting. In the Meeting Review, Christof Sparr summarizes the program of the 2015 conference.
Structures, Reactions, and Mechanisms: Stereochemistry in the Broadest Sense at the 51st Bürgenstock Conference
- First Published: 23 June 2016

Tradition and innovation: The 51st Bürgenstock Conference on Stereochemistry took place from May 1–6, 2016 and offered its usual mixture of impressive science and superb discussions in a wonderful atmosphere. In the Meeting Review, Ivana Fleischer outlines the program.
Form Follows Function: Designer Chemistry at the 52nd Bürgenstock Conference
- First Published: 04 July 2017

The 52nd Bürgenstock Conference on Stereochemistry took place from April 30–May 4, 2017, and showed how chemistry and design go hand-in-hand (as reflected in the image of the Bauhausarchiv in Berlin). In this Conference Report, Philipp Heretsch outlines the program.
Merging Art and Science—The 53rd Bürgenstock Conference
- First Published: 13 July 2018

For the 53rd time, the Bürgenstock Conference gathered some of the most gifted scientists and rising stars in organic, physical, and bioorganic chemistry. Orchestrated by Ilan Marek (President) and his successor, Véronique Gouverneur, the synergy between art and science took place in Brunnen, Switzerland, with a beatiful view over Lake Lucerne.
Guest of Honor
Spectroscopic Observation of the Triplet Diradical State of a Cyclobutadiene
- First Published: 21 June 2017

Squaring the rectangle: Thermal excitation of the cyclobutadiene singlet 1 leads to the first direct detection by EPR spectroscopy of a triplet diradical state of a cyclobutadiene derivative 2. Both experiment and theory support a thermal equilibrium between rectangular singlet 1 and the square triplet diradical 2 with a singlet–triplet energy gap of 13.9 kcal mol−1.
Organizing Committee/Moderators
Supramolecular Copolymerization as a Strategy to Control the Stability of Self-Assembled Nanofibers
- First Published: 02 May 2018
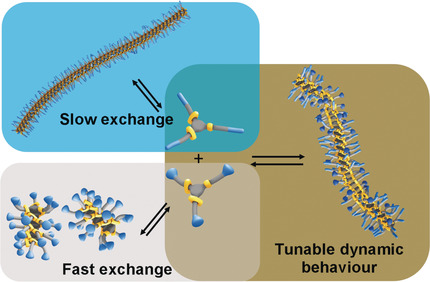
The exchange dynamics of supramolecular polymers based on benzene-1,3,5-tricarboxamide (BTA) can be regulated by copolymerizing molecules with dendronized (dBTA) and linear (nBTA) ethylene glycol-based water-soluble side chains. Whereas nBTAs form long nanofibers in water, dBTAs do not polymerize. The copolymerization of the two BTAs results in more stable long nanofibers.
Palladium-Catalyzed Decarbonylative Trifluoromethylation of Acid Fluorides
- First Published: 25 February 2018
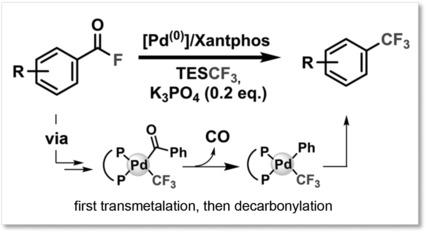
Homogeneous catalysis: Trifluoromethyl arenes have been obtained by Pd-catalyzed decarbonylative functionalization of acid fluorides. The key PdII-F intermediate forms through intramolecular redistribution to Pd, allowing direct transmetalation to occur. Computational and experimental mechanistic studies reveal that transmetalation occurs prior to decarbonylation.
Lewis Acid Catalyzed Enantioselective Desymmetrization of Donor–Acceptor meso-Diaminocyclopropanes
- First Published: 20 February 2018
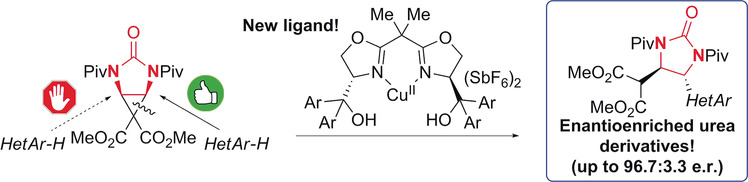
Open carefully: The Lewis acid catalyzed enantioselective ring-opening desymmetrization of a donor–acceptor meso-diaminocyclopropane is enabled by a copper(II) complex bearing a novel BOX ligand. The Friedel–Crafts alkylation of indoles and one pyrrole with this new meso-diaminocyclopropane delivers diastereomerically pure and highly enantioenriched urea derivatives.
Rhodium-Catalyzed Arylation of Cyclopropenes Based on Asymmetric Direct Functionalization of Three-Membered Carbocycles
- First Published: 15 February 2018

Small ′Rh′ings: The asymmetric rhodium-catalyzed arylation of simple achiral nonfunctionalized cyclopropenes provides a variety of arylated cyclopropanes with high diastereo- and enantioselectivities (e.r. from 90:10 to 99:1). The reaction proceeds under mild reaction conditions. cod=1,5-cyclooctadiene.
The First Gold(III) Formate: Evidence for β-Hydride Elimination
- First Published: 13 September 2017

An anionic ligand exchange reaction on [(N^C^C)AuIIIF] in the presence of formic acid delivered the first example of a stable gold(III) formate. Its ability to undergo β-hydride elimination and its reactivity in the dehydrogenation of formic acid (FA) have been experimentally and computationally demonstrated.
Copper-Catalyzed Insertion into Heteroatom–Hydrogen Bonds with Trifluorodiazoalkanes
- First Published: 16 February 2016
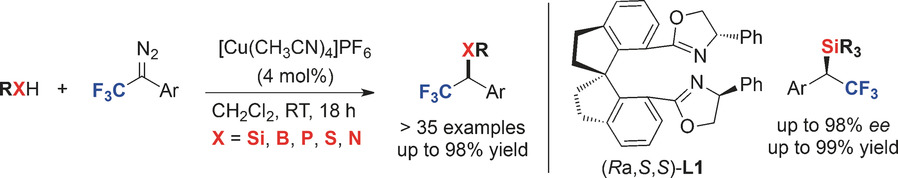
Suits all sorts: An efficient copper-catalyzed carbenoid insertion of 2,2,2-trifluorodiazoethane and 1-aryl 2,2,2-trifluorodiazoethanes into Si−H, B−H, P−H, S−H, and N−H bonds produced CF3-containing products in high yields (see scheme). Catalytic asymmetric Si−H and B−H bond insertion reactions were also developed with chiral bisoxazoline ligands, such as (Ra,S,S)-L1.
Speakers
Single-Molecule Determination of the Isomers of d-Glucose and d-Fructose that Bind to Boronic Acids
- First Published: 24 January 2018
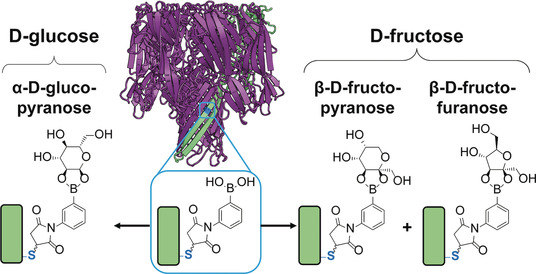
Sweet reaction: The aqueous, reversible covalent chemistry of boronic acids and diols can be monitored inside a protein nanoreactor. The approach is used to identify which cyclic isomers of d-glucose and d-fructose bind to a boronic acid in aqueous solution. Both of these binding modes contradict current binding models.
Quantitative and Comparative Profiling of Protease Substrates through a Genetically Encoded Multifunctional Photocrosslinker
- First Published: 20 September 2017
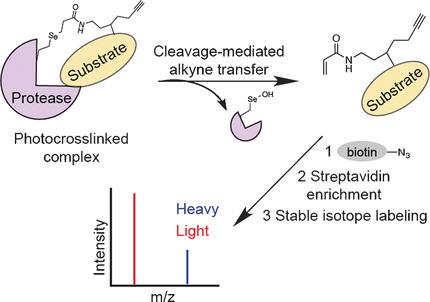
A genetically encoded trifunctional photocrosslinker was developed that bears a bioorthogonal handle and a releasable linker in addition to its photoaffinity warhead. This probe enabled the enrichment of transient and low-abundance prey proteins after intracellular photocrosslinking and prey–bait separation. A quantitative and comparative proteomic strategy using this probe revealed the substrates of a protease.
Affinity Enhancement of Protein Ligands by Reversible Covalent Modification of Neighboring Lysine Residues
- First Published: 06 November 2018
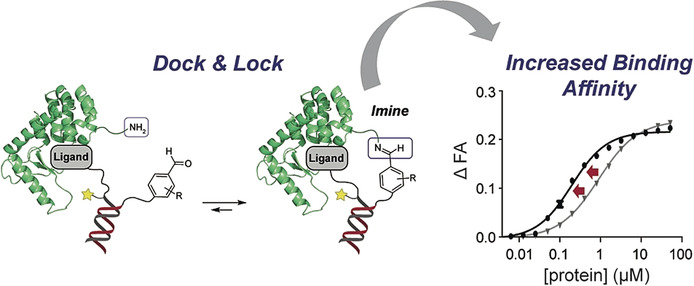
A lock on affinity: The simultaneous display of protein ligands and of 2-hydroxybenzaldehyde derivatives on either two complementary LNA strands or a small-organic molecule leads to an enhanced binding affinity by reversible covalent interaction with proximal lysine residues.
Design of Photostable, Activatable Near-Infrared Photoacoustic Probes Using Tautomeric Benziphthalocyanine as a Platform
- First Published: 07 April 2019
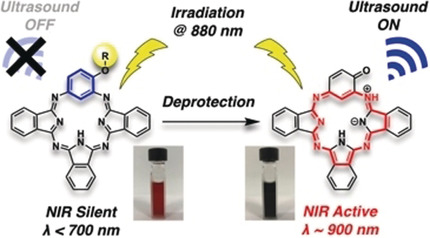
Photoacoustic imaging: An activatable near-infrared (NIR) system based on tautomeric benziphthalocyanines (BPcs) was developed for photoacoustic imaging. These water-soluble hydroxy BPcs exhibit high photostability and no fluorescence. The photoacoustic signals of the hydroxy-masked BPcs increase upon near-infrared excitation at 880 nm in the presence of esterase or H2O2.
The Cation–π Interaction in Small-Molecule Catalysis
- First Published: 22 June 2016
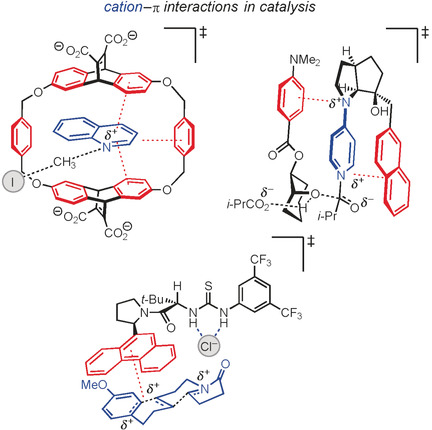
Positive effects: This Review discusses the burgeoning role of the cation–π interaction in the small-molecule catalysis of organic and organometallic reactions. An extensive survey of the state-of-the-art research emphasizes how unexpected discoveries and systematic mechanistic studies have begun to enable rational applications of the cation–π interaction in catalyst design.
Asymmetric and Geometry-Selective α-Alkenylation of α-Amino Acids
- First Published: 02 January 2019
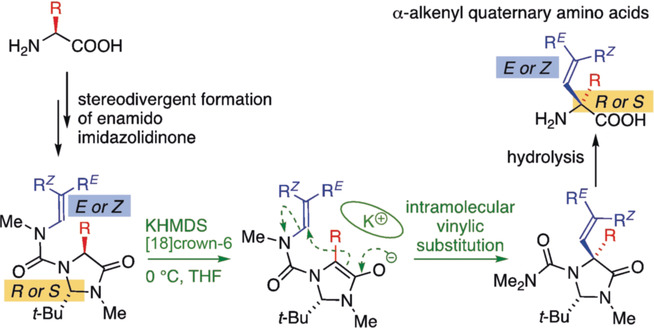
Stereocontrol: E- and Z-N′-alkenyl urea derivatives of imidazolidinones may be formed selectively from enantiopure α-amino acids. Generation of their enolate derivatives in the presence of K+ and [18]crown-6 induces intramolecular migration of the alkenyl group from N′ to Cα. Hydrolysis of the enantiopure products under acid conditions reveals quaternary α-alkenyl amino acids.
Sign up for email alerts
Tools
A Journal of the
German Chemical Society
More from this journal
Classifieds: Jobs and Awards, Products and Services
- Angewandte Chemie International Edition
- 4 November 2024
Graphical Abstract

Are you looking for a (new) job? Do you know someone who deserves an award? Are you trying to find a product or service to make your work more efficient? Our partners can help.
Related Titles



 IssueVolume 97, Issue 7
IssueVolume 97, Issue 7Special Issue: Membrantechnik
July 2025Guest Editor(s):
Deutsche Gesellschaft für Membrantechnik