Journal list menu

 Issue
IssueAngewandte Chemie International Edition in English: Volume 27, Issue 8
1009-1112August 1988
Export Citations
Download PDFs
Cover Picture (Angew. Chem. Int. Ed. Engl. 8/1988)
- First Published: August 1988
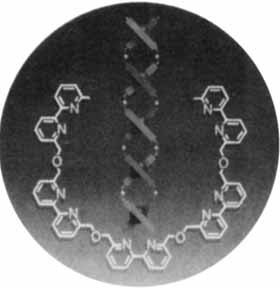
The cover picture shows a schematic example of molecular self-organization. Simple double-helical complexes, so-called helicates, form spontaneously from a penta(bipyridine) ligand and Cu1 ions in solution. Whereas hydrogen bonds and stacking interactions are responsible for holding together the double helix of natural DNA, coordination interactions between Cu⊕ and bipyridine units play the key role in determining the structure of these rudimentary, double-stranded molecules. Further lengthening of the ligands should lead to self-organizing nanostructures. The field is wide open for organic, inorganic, physical, and biochemical studies: modifications of the subunit; complexation of other metal ions; investigations of binding cooperativity, of the dynamics of double helix formation and unraveling, and of the interaction with DNA; construction of self-organizing and self-amplifying devices. Further details on helicates are reported by J.-M. Lehn et al. on p. 1095 f.
Graphical Abstract (Angew. Chem. Int. Ed. Engl. 8/1988)
- First Published: August 1988
Reviews
The Design of Molecular Hosts, Guests, and Their Complexes (Nobel Lecture)†
- Pages: 1009-1020
- First Published: August 1988
The founders of “supramolecular chemistry” include Charles J. Pedersen, Donald J. Cram, and Jean-Marie Lehn, who shared the Nobel Prize for Chemistry in 1987 for their fundamental research in this area of organic chemistry. Their work focused the attention of many chemists on the cavities formed by certain types of molecules. Cations, anions, or neutral molecules can enter the cavities of specifically designed compounds and are held there by intermolecular forces. It is fully justified, therefore, to compare such compounds to biomolecules. How the development began, how it achieved its first successes, and what fascinating possibilities lie in store for future research are discussed by Pedersen and Cram in this issue and by Lehn in the January issue (page 89 ff.). The most recent results from Lehn's research are also reported, appropriately, in this issue.
The Discovery of Crown Ethers (Noble Lecture)†
- Pages: 1021-1027
- First Published: August 1988
The founders of “supramolecular chemistry” include Charles J. Pedersen, Donald J. Cram, and Jean-Marie Lehn, who shared the Nobel Prize for Chemistry in 1987 for their fundamental research in this area of organic chemistry. Their work focused the attention of many chemists on the cavities formed by certain types of molecules. Cations, anions, or neutral molecules can enter the cavities of specifically designed compounds and are held there by inter-molecular forces. It is fully justified, therefore, to compare such compounds to biomolecules. How the development began, how it achieved its first successes, and what fascinating possibilities lie in store for future research are discussed by Pedersen and Cram in this issue and by Lehn in the January issue (page 89 ff.). The most recent results from Lehn's research are also reported, appropriately, in this issue.
Somatic Generation of Immune Diversity (Nobel Lecture)†
- Pages: 1028-1039
- First Published: August 1988
How does an organism manage during its lifetime to respond to a huge number of different antigens, even though the number of antibody genes is, of necessity, limited? The Nobel Prize for Medicine was awarded to Susumu Tonegawa in 1987 for providing an answer to this question. Tonegawa discovered that not a single complete gene for an antibody peptide chain is inherited by an organism. Instead, the genetic information in the germ line is contained in several hundred gene segments. Somatic recombination of these gene segments during the differentiation of B lymphocytes results in the production of tens of thousands of complete genes. Somatic hypermutation of these genes leads to further diversification of antibody peptide chains in such a way that B cells that carry immunoglobulin receptors displaying good recognition of specific antigens are selected for in a later phase of B-cell differentiation.
Protein Phosphorylation in Bacteria—Regulation of Gene Expression, Transport Functions, and Metabolic Processes
- Pages: 1040-1049
- First Published: August 1988
The reversible phosphorylation of proteins is responsible for controlling a large number of biological processes—not only in eukaryotic cells but also in bacteria. For example, the phosphorylation of isocitrate dehydrogenase determines, under certain conditions of growth, whether isocitrate is degraded via the citric acid cycle or the glyoxylate pathway. In such processes, the phosphate group becomes attached to the amino acids serine, threonine, tyrosine, and histidine.
Say It in English, Please!†
- Pages: 1050-1057
- First Published: August 1988
“… I don't set out to teach, but to entertain. I hope to give you a feeling for the English language, and to communicate some of my love for it.” What better way to describe the purpose behind these nine short essay! They originally constituted the first installments in a series of articles aimed at German-speaking writers of English.
Communications
Triple Proton Abstraction from a 1-Methylallyl Ligand on Thermolysis: Formation of a Hf3(C4H4) Cluster†
- Pages: 1058-1059
- First Published: August 1988
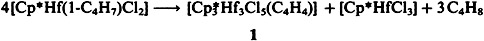
Complexes containing the unsubstituted 1,3-butadiene-1,4-diyl ligand C4H4 are very rare. Such a complex, 1, has now been generated, surprisingly, by the unusual title reaction. In 1, the C4H4 ligand is linked unsymmetrically to three Cp*ClHf moieties; the two remaining Cl atoms each bridge two Hf centers (Cp* = η5-C5Me5).
Alkali-Metal Hydrogen Tetraphosphides, MIHP4—the First Salts of Bicyclo[1.1.0]tetraphosphane†‡
- Pages: 1059-1061
- First Published: August 1988
![Alkali-Metal Hydrogen Tetraphosphides, MIHP4—the First Salts of Bicyclo[1.1.0]tetraphosphane](/cms/asset/bfe16190-49a7-415e-9b6f-dc3de41866f5/must001.jpg)
The first isolable reduction products of white phosphorus, the title compounds, are formed upon reaction of P4 with naphthalenelithium or -sodium/potassium in protic solvents. They contain the HP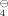 ion 1, which, according to its NMR data, displays an unusual electronic structure. Intramolecular exchange of the endo hydrogen atom does not occur.
ion 1, which, according to its NMR data, displays an unusual electronic structure. Intramolecular exchange of the endo hydrogen atom does not occur.
Synthesis and Physical Properties of 3,6-Di-tert-butyl-1,4-diazapentalene
- Pages: 1061-1062
- First Published: August 1988
The pentalene derivative 2 is only inductively stabilized. It was obtained by reaction of the dihydro derivative 1 with nickel peroxide. Comparison of the spectroscopic data with the results of MNDO calculations indicates that the Kekulé structure shown here dominates; that is, the bonds are fixed.
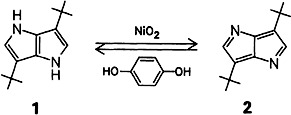
Catalytic Isomerization of Methylated 1,5-Cyclooctadienes†‡
- Pages: 1062-1064
- First Published: August 1988
Methyl substituents—and the temperature—influence the NiII-catalyzed isomerization of 1,5-cyclooctadienes. For example, reaction of isoprene in refluxing hexane leads, via 1, to the bicyclic compound 2 and reaction of isoprene and trans-piperylene leads, via 3, to the highly strained tricyclic cyclopropane derivative 4. The structures of the carbon frameworks were established by 2D INADEQUATE NMR experiments.
Strongly Bent Naphthalene Units in [2](2,6)Naphthalino[2]paracyclophane and [2](2,6)Naphthalino[2]paracyclophane-1, 11-diene
- Pages: 1064-1065
- First Published: August 1988
Naphthalino[2]paracyclophane and [2](2,6)Naphthalino[2]paracyclophane-1, 11-diene](/cms/asset/45556043-11e0-4dbb-900d-64a611bec97f/must001.jpg)
Fused-ring arenes such as naphthalene can be subjected to considerable deformation. This is exemplified by the structures of the two title compounds. They also provide further support for the suggested stability of such molecules as the three-dimensional, sphere-shaped cluster C60 (“Buckminsterfullerene” or “Footballene”), which consists of 60 sp2-hybridized carbon atoms. The phane-diene is shown on the right.
Kinetic and Stereochemistry of the SO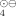 -Induced Hydroxylation of Cyclohexenes in Aqueous Solution
-Induced Hydroxylation of Cyclohexenes in Aqueous Solution
- Pages: 1066-1067
- First Published: August 1988
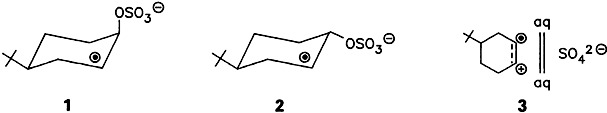
A largely solvent-separated ion pair 3 is the presumed intermediate in the hydroxylation of cyclohexenes with SO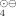 . Radical anions like 1 and 2 are formed in a nearly diffusion-controlled step. They are subsequently hydrolyzed via 3. The axial/equatorial selectivity of the reaction of H2 O with 3 is much greater than that observed in the reaction of OH⊙ with 4-tert-butylcyclohexene (90:10 vs. 58:42).
. Radical anions like 1 and 2 are formed in a nearly diffusion-controlled step. They are subsequently hydrolyzed via 3. The axial/equatorial selectivity of the reaction of H2 O with 3 is much greater than that observed in the reaction of OH⊙ with 4-tert-butylcyclohexene (90:10 vs. 58:42).
Isolation and Structure of [(Fluorenone ⊙⊖){Na⊕(dme)2}]2—a Touchstone for Radical Contact Ion Pairs?†‡
- Pages: 1067-1069
- First Published: August 1988
Single crystals of a radical ion pair salt—the title compound—could be grown after stoichiometric reduction of fluorenone with sodium in aprotic solution under argon at –50°C. The structure determination reveals that a dimeric contact ion pair containing an Na2O2 four-membered ring is present and shows, for the first time, the bonding between radical anions and countercations: The solvated Na⊕ ion is situated above the plane of the molecule and outside of the πCO plane. This finding further supports the assumption of a π spin delocalization from the radical anion to the metal cation, as derived from the positive ENDOR metal couplings of radical ion pairs in solution.
ENDOR Spectroscopy in Water: Reductions and Oxidations of Organic Compounds†‡
- Pages: 1069-1071
- First Published: August 1988
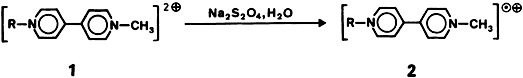
The characterization of paramagnetic compounds in water by ENDOR spectroscopy was achieved by the use of special capillary tubes and optimal concentrations. This extension of the scope of application of this method is of great value for the study of biological redox reactions. The method was exemplified by determining the spectrum of the radical cation 2, R = n-dodecyl. This compound was obtained by reduction of the viologen 1.
Dications Based on a Hydrotris(phosphonio)borate Skeleton†
- Pages: 1071-1074
- First Published: August 1988
The first two dications having a [HBP3]2 ⊕ framework (1 and 2)—the boron atom is surrounded by three phosphonium centers and a hydrogen atom—were isolated as dibromides and characterized by X-ray structure analysis. The stability of the two salts toward air and moisture is remarkable. The boron-bonded H atom is exchanged for a D atom in alkaline D2O.
From the Dilithiooctamethylcyclotetrasilazane to the 1,3,5,7-Tetraaza-2,4,6,8,9-pentasilabicyclo[3.3.1]nonane System†‡
- Pages: 1074-1075
- First Published: August 1988
![From the Dilithiooctamethylcyclotetrasilazane to the 1,3,5,7-Tetraaza-2,4,6,8,9-pentasilabicyclo[3.3.1]nonane System](/cms/asset/c1f10b72-f097-4278-8b00-854155d7bad9/must001.jpg)
Reaction of the long-known cyclotetrasilzane derivative 1 was accomplished for the first time without ring-opening, namely by treatment with nBuLi to afford the dimer [2]2, in which two eight-membered rings are joined through an (LiN)2 four-membered ring. Depending on the molar ratios used, reaction of 2 with PhSiF3 results in either disubstitution or bridging to give 3.
Azirineimine as a Structural Unit in the Side Chain of a Hexofuranose†
- Pages: 1075-1077
- First Published: August 1988
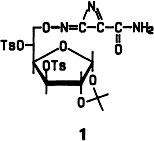
Azirineimine, formally an HCN dimer, was obtained for the first time as derivative 1. Compound 1 is formed upon reaction of 1,2-O-isopropylidene-3,5,6-tri-O-p-toluenesulfonyl-α-D-glucose with KCN in nitromethane/water under phase-transfer conditions. Theoretical studies have shown that azirineimine may have played a role in the prebiotic formation of organic compounds.
Intramolecularly Stabilized Organogallium Compounds†
- Pages: 1077-1078
- First Published: August 1988
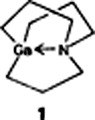
Compound 1 has an azagallapropellane structure in which the GaR3 unit is stabilized intramolecularly by the N atom. Ga has a trigonal-pyramidal environment. Compound 1 is formed from GaCl3 and the Grignard derivative of tris(3,3′,3″-chloropropyl)amine. Despite the coordinatively unsaturated pyramidal base, 1 is remarkably stable, for example toward oxygen.
Tris(thioxanthen-9-ylidene)cyclopropane, and Its Radical Cation and Dication†
- Pages: 1078-1080
- First Published: August 1988
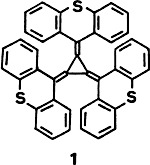
A [3]radialene having strong electron-donor properties, the title compound 1, was formed upon reaction of lithium thioxanthenide with hexachlorocyclopropane after addition of BuLi. Cyclic voltammetry in CH2Cl2 gave two pairs of reversible waves corresponding to the formation of 1⊙⊕ and 12⊕. A thermally accessible triplet state, only 0.07 eV higher in energy than the singlet state, was established for dark blue 12⊕·2CF3CO by ESR spectroscopy. Thus, 1 might serve as a donor component for the preparation of organic ferromagnets.
by ESR spectroscopy. Thus, 1 might serve as a donor component for the preparation of organic ferromagnets.
Tris(fluoren-9-ylidene)cyclopropane, a Novel [3]Radialene†
- Pages: 1080-1081
- First Published: August 1988
![Tris(fluoren-9-ylidene)cyclopropane, a Novel [3]Radialene](/cms/asset/fa039e06-7c66-4e5a-b006-e53321f8284d/must001.jpg)
NMR data show that the title compound 2 has a highly symmetrical structure—a propeller-like arrangement having D3 symmetry is plausible. Compound 2 was prepared by cyclotrimerization of the ate complex 1. An explanation for the easy, reversible, two-step reduction of 2 to its dianion may be provided by the importance of the dipolar resonance structure 3 in describing the actual structure of the dianion.
Is Tetrasilatetrahedrane Kinetically Stable?†
- Pages: 1081-1083
- First Published: August 1988
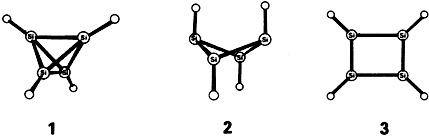
The answer is probably no, even though it violates common expectations. Calculations on 1–3, taking into consideration electron correlation, gave 1 as the highest-energy species at all levels of approximation. This reflects once again the fact that—in contrast to the carbon analogues—Si3 rings are more highly strained than Si4 rings.
1,2-Dilithioethane from Ethylene and Lithium
- Pages: 1083-1084
- First Published: August 1988
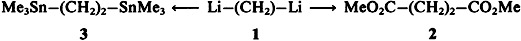
Generation of the thermally unstable 1,2-dilithioethane 1, which had eluded earlier synthetic attempts, has now been carried out under special conditions. Condensation of lithium vapor on a glass formed from ethylene and dimethyl ether at — 196°C yielded highly active lithium, which, upon melting of the glass, reacted with ethylene to afford 1. Compound 1 was identified by carboxylation and methylation to give the ester 2 and by stannylation to give the bis(stannane) 3.
The Activation of Phenylmagnesium Compounds by Crown Ethers†
- Pages: 1084-1086
- First Published: August 1988
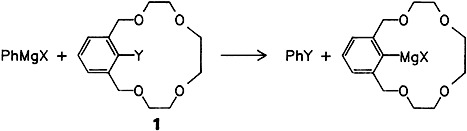
“Organolithium-like” behavior of phenylmagnesium compounds is possible to achieve using the activating influence of the crown ether derivative 1. Halogen-metal exchange and metalation were previously considered to be reactions characteristic of organolithium (but not organomagnesium) compounds. The new reactions are also of preparative interest (X = Ph, Br; Y = H, Br).
β-Mannosides from β-Glucosides by Intramolecular Nucleophilic Substitution with Inversion of Configuration†
- Pages: 1086-1087
- First Published: August 1988
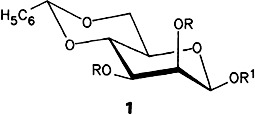
The β-mannoside bond in the core region of N-glycoproteins is difficult to form. The epimerization at C-2 of β-glucosides, which is normally difficult to achieve, has now been accomplished by intramolecular nucleophilic attack of a carbamate substituent in the sense of neighboring-group assistance. It was possible in this way to prepare directly isomerically pure (protected) β-mannosides like 1 from menthol and cholesterol as well as β-mannosidic disaccharides.
Polycyclic Silylamides of GeII and SnII with Different Structures—Bis(germanediyl) versus Distannate†
- Pages: 1087-1089
- First Published: August 1988
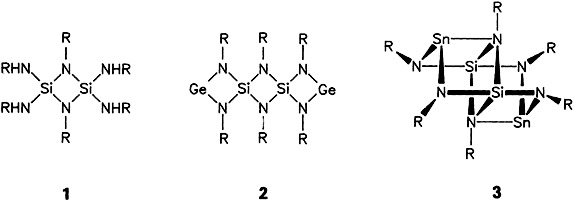
The same educt, homologous reagents, products having analogous empirical formulas—yet totally different structures! Whereas Ge11 is incorporated into the cyclodisilazane 1 to form the bis(germanediyl) 2 containing divalent Ge, 1 reacts with Sn11 to form the stannate 3 containing trivalent Sn. These results are ascribed to ring-strain effects (R = tBu).
The Enzymic Interconversion of Isobutyryl and n-Butyrylcarba(dethia)-Coenzyme A: A Coenzyme-B12-dependent Carbon Skeleton Rearrangement†‡
- Pages: 1089-1090
- First Published: August 1988
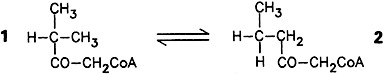
The synthetic carba(dethia) analogues of coenzyme A contain a —CO—CH2—instead of a —CO—S— group and are therefore hydrolytically stable. The isobutyrylcarba(dethia) coenzyme A 1, whose synthesis is reported here, is isomerized to the n-butyryl derivative 2 by a cell extract from Streptomyces cinnamonensis (monitored by 1H NMR). This new, coenzyme B12-dependent structural rearrangement is similar in many ways to the long-known methylmalonyl-CoA mutase reaction.
Isolated, Complex Zintl Molecules in the Gas Phase†
- Pages: 1091-1092
- First Published: August 1988
Ternary and quaternary intermetallic molecules such as Cs2InSb3 and CsSnBiSb2, respectively, can be detected in the vapor above the corresponding alloys by mass spectrometry. The composition of the compounds is in agreement with the Zintl-Klemm-Busmann concept, if one assumes that the alkali metal transfers an appropriate number of electrons to the elements of the third and fourth main groups in the Sb4 tetrahedra, such that these elements become isoelectronic to Sb.
A Monomeric Potassium Salt of Bis(trimethylsilyl)methyl Phenyl (Sulfone and the Determination of the 1J(13C,29Si) Coupling Constants of Si-Stabilized “Carbanions”†‡
- Pages: 1092-1094
- First Published: August 1988
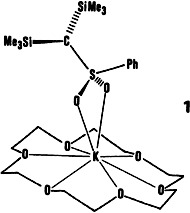
Monomeric metalated sulfones like 1 are a rarity. In 1, K⊕ is bonded only to the O atoms of the sulfonyl group. Accordingly, the negative charge is stabilized largely by polarization and negative anionic hyperconjugation. Comparison of the effect of metalation on the 1J(13C, 29Si) values of several carbanions stabilized by Si (and S) showed that these coupling constants are good probes for the structure of carbanions in solution.
Photolytic Cluster Synthesis: Synthesis and Structure of [{(Ph3P)Au}7Mo(CO)3]OH†
- Pages: 1094-1095
- First Published: August 1988
The photolytic elimination of N2 from [(Ph3P)AuN3] is the basis of a new method for preparing gold clusters. The photolysis of a mixture of [(Ph3P)AuN3] and [Mo(CO)6] in THF results in loss of N2 and CO and formation of the title compound, in which the Mo atom forms the center of a novel icosahedral fragment consisting of seven Au atoms. Only a few examples of gold clusters containing heterometal centers were previously known.
Helicates: Tetra- and Pentanuclear Double Helix Complexes of CuI and Poly(bipyridine) Strands†‡
- Pages: 1095-1097
- First Published: August 1988
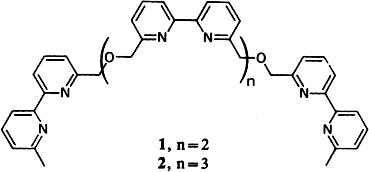
Inorganic double helices having 2 and 2.5 turns are formed by the tetra- and penta(bipyridine) ligands 1 and 2, respectively, upon addition of Cu1 salts. These polynuclear complexes, in which the copper ions are aligned like a string of pearls, are estimated to be about 22 and 27 nm long, respectively.
Methyl 4-Hydroxy-5-iodo-2,3-dimethoxy-6-methylbenzoate: The Aromatic Fragment of Calichemicin γ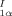 . Synthesis, X-Ray Crystallographic Analysis, and Properties†
. Synthesis, X-Ray Crystallographic Analysis, and Properties†
- Pages: 1097-1099
- First Published: August 1988
The biologically highly active calichemicin γ contains the benzoate 1 — glycosylated on the OH group and linked through the ester group (S instead of O), via sugar moieties, to other unusual organic fragments. Compound 1 has now been synthesized in five steps from 3,4,5-trimethoxytoluene and has proven to be very interesting. It undergoes spontaneous enantiomer resolution upon crystallization and exhibits second harmonic generation activity.
contains the benzoate 1 — glycosylated on the OH group and linked through the ester group (S instead of O), via sugar moieties, to other unusual organic fragments. Compound 1 has now been synthesized in five steps from 3,4,5-trimethoxytoluene and has proven to be very interesting. It undergoes spontaneous enantiomer resolution upon crystallization and exhibits second harmonic generation activity.
Pentadienyl, a More Reactive and More Strongly Bound Ligand Than Cyclopentadienyl†
- Pages: 1099-1101
- First Published: August 1988
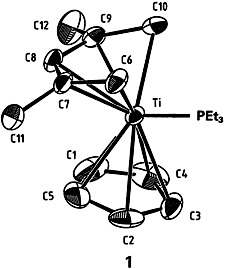
Direct comparison of the cyclopentadienyl ligand with its open-chain counterpart has been accomplished using complex 1, which contains both ligands. The Ti-C bonds to the acyclic ligand are 0.106 Å shorter than those to the cyclic ligand. Although this finding indicates that the bonds to the pentadienyl ligand are stronger, reaction of 1 with acetonitrile occurs nonetheless at this ligand. The aromatic character of C5H offers a possible explanation for this apparent contradiction. The aromaticity results in resistance to chemical change; that is, both formation of stronger bonds and coupling with electrophiles are disfavored.
offers a possible explanation for this apparent contradiction. The aromaticity results in resistance to chemical change; that is, both formation of stronger bonds and coupling with electrophiles are disfavored.
Phase Transformations of Lithium Nitride under Pressure
- Pages: 1101-1103
- First Published: August 1988
The transformation of α-Li3N to β-Li3N occurs at 600 MPa and room temperature. The reverse transformation, however, only begins to take place above 200°C under normal pressure. β-Li3N crystallizes as the Na3As structure type. This modification allows Li3N to be placed systematically in the structural series of alkali-metal compounds A3B involving elements of the nitrogen group (A = Li, Na, K, Rb, Cs; B = N, P, As, Sb, Bi). Since the phase transition occurs at relatively low pressure, the formation of small amounts of β-Li3N upon grinding with a mortar and pestle cannot be ruled out. Indeed, X-ray powder diffractions of α-Li3N always reveal the presence of small amounts of β-Li3N.
Functionalized, Enantiomerically Pure [2.1.1]-, [2.2.1]-, and [2.2.2]-Triblattanes†
- Pages: 1103-1106
- First Published: August 1988
![Functionalized, Enantiomerically Pure [2.1.1]-, [2.2.1]-, and [2.2.2]-Triblattanes](/cms/asset/a60518ad-fbfd-462b-971d-d12c9e4eb29a/must001.jpg)
Triblattanes, such as the previously known 1—(+)-1 is shown here—are very promising, novel cage molecules. For example, 1 can be transformed efficiently into [2.2.2]-triblattatriene (+)-/(-)-2, a C14H14 pentacyclic compound, by means of a threefold ring expansion. Like its similarly obtained tribenzo derivative, 2 has D3 symmetry. The chemical versatility of 2 and its precursors is exemplified by the generation of the hexaketone 3, which was trapped as its triquinoxaline derivative.
Book Reviews
Book Review: Organic Chemistry—in Color, and Good: Organic Chemistry. By K. P. C. Vollhardt
- Pages: 1106-1107
- First Published: August 1988
Book Review: Organic Chemistry—in Color, and Good: The Chemistry of Inorganic Homo- and Heterocycles. Vol. 1 and 2. Edited by I. Haiduc and D. B. Sowerby
- Pages: 1107-1108
- First Published: August 1988
Book Review: Organic Chemistry—in Color, and Good: Industrielle Aromatenchemie. Rohstoffe, Verfahren, Produkte. By H.-G. Franck and J. W. Stadelhofer
- Pages: 1108-1109
- First Published: August 1988
Book Review: Organic Chemistry—in Color, and Good: Advances in Electrophoresis. Vol. 1. Edited by A. Chrambach, M. J. Dunn and B. J. Radola
- Pages: 1109-1110
- First Published: August 1988
Book Review: Organic Chemistry—in Color, and Good: Protein Engineering. Edited by D. L. Oxender and C. F. Fox
- Page: 1110
- First Published: August 1988
Book Review: Organic Chemistry—in Color, and Good: The Chemistry of the Metal-Carbon Bond. Vol. 4: The Use of Organometallic Compounds in Organic Synthesis. Edited by F. R. Hartley
- Pages: 1110-1111
- First Published: August 1988
Book Review: Organic Chemistry—in Color, and Good: The Chemistry of the Actinide Elements. 2nd Edition. Edited by J. J. Katz, G. T. Seaborg, and L. R. Morss
- Pages: 1111-1112
- First Published: August 1988
Tools
A Journal of the
German Chemical Society
More from this journal
Classifieds: Jobs and Awards, Products and Services
- Angewandte Chemie International Edition
- 4 November 2024
Graphical Abstract

Are you looking for a (new) job? Do you know someone who deserves an award? Are you trying to find a product or service to make your work more efficient? Our partners can help.
Related Titles



 IssueVolume 97, Issue 7
IssueVolume 97, Issue 7Special Issue: Membrantechnik
July 2025Guest Editor(s):
Deutsche Gesellschaft für Membrantechnik