

Journal list menu

 Issue
IssueJournal of Computational Chemistry: Volume 46, Issue 1
January 5, 2025
Export Citations
Download PDFs
COVER IMAGE

ISSUE INFORMATION
SHORT COMMUNICATION
Efficient acceleration of the convergence of the minimum free energy path via a path-planning generated initial guess
- First Published: 18 September 2024
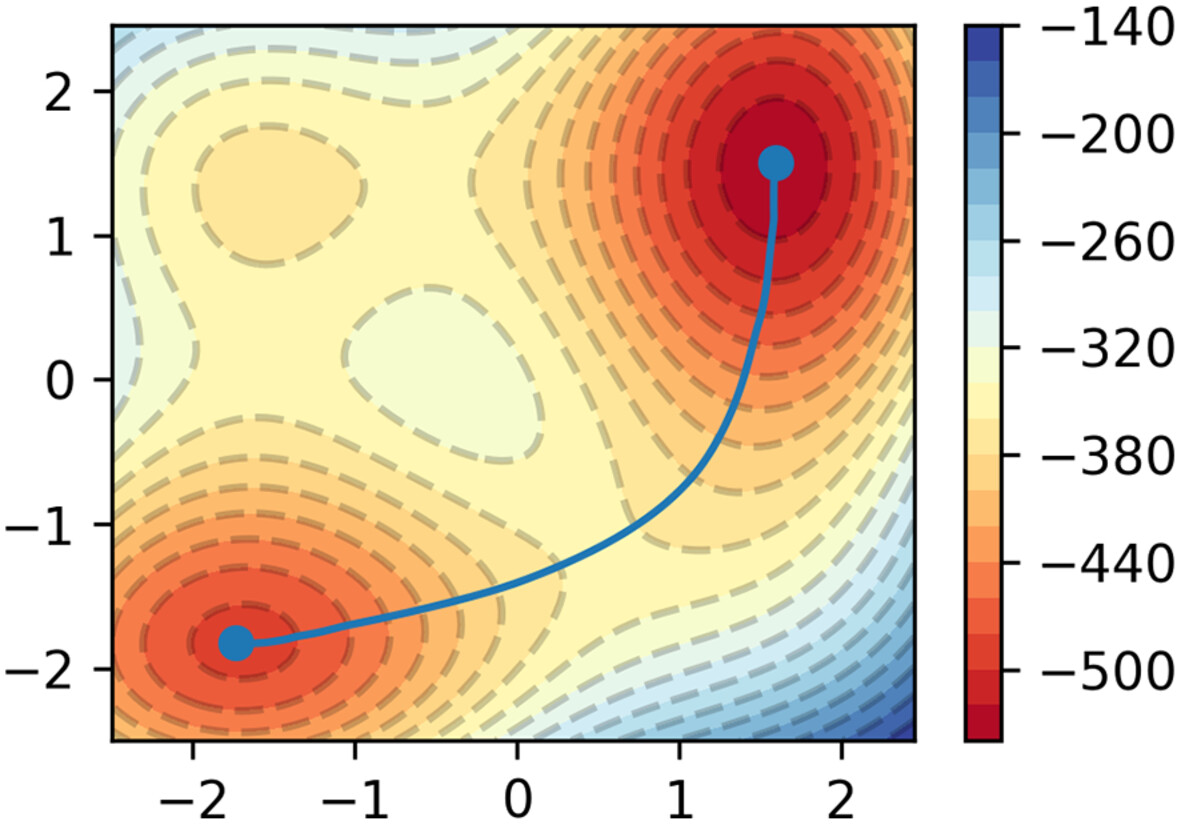
This work employs a combined fast marching tree (FMT)-string algorithm to determine the minimum energy path (MEP). The idea behind this method is that using FMT can give the MEP good initial approximations, so only a few iterations are required for refinement. This saves time and the number of iterations required compared to using a linear-interpolated (LI) initial guess. After that, we show that this method works in a number of model free energy surfaces (FES), including the well-known Muller-Brown potential, where FMT-string is better than the LI counterpart at getting a converged MEP. We also explore the potential applications of this method in molecular systems.
RESEARCH ARTICLE
Modeling the effect of substituents on the electronically excited states of indole derivatives
- First Published: 20 September 2024
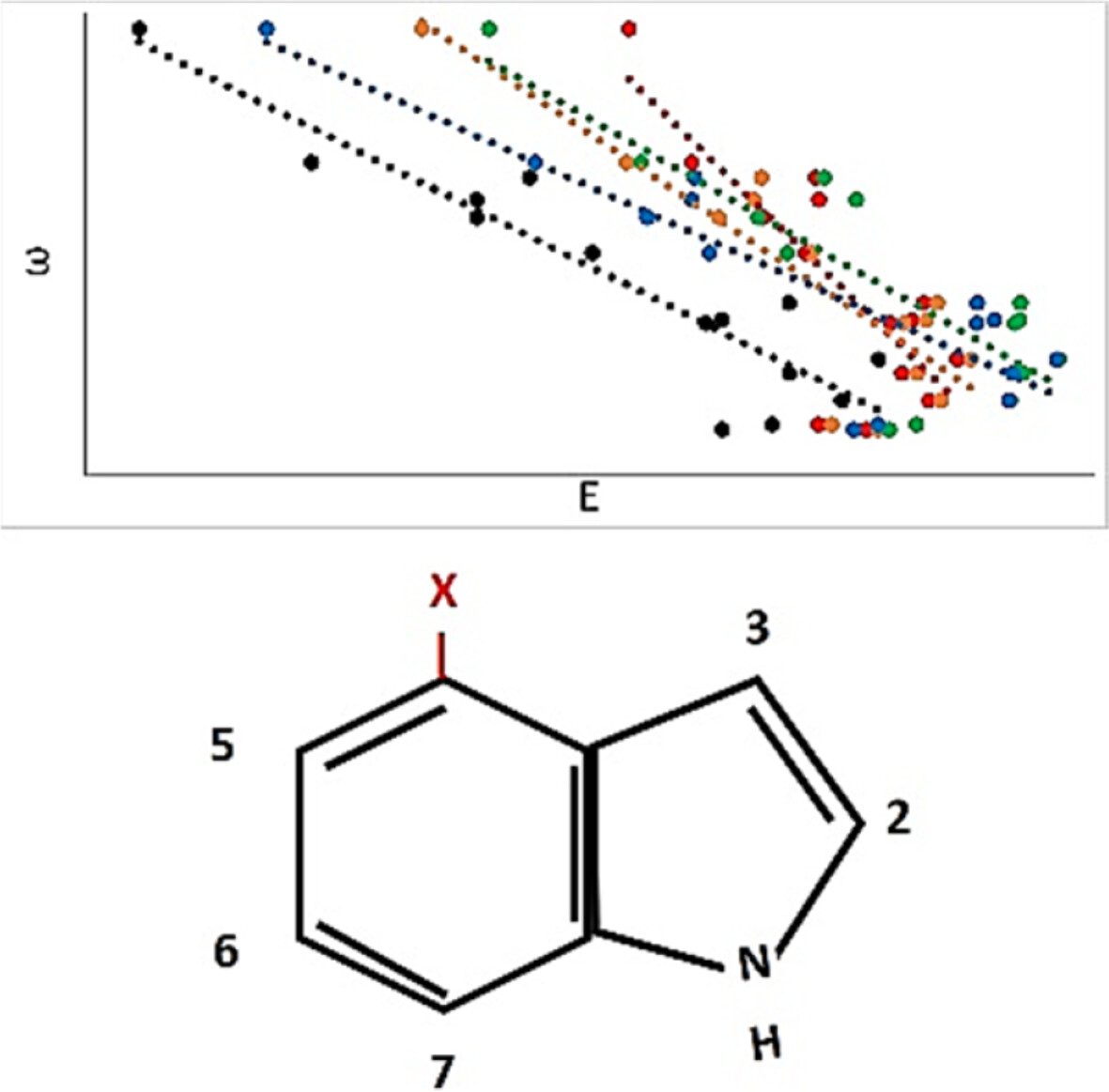
Excited state calculations were performed on fourth-position indole derivatives to evaluate substituent effects on indole's absorption. Results show correlation of excitation energies to the electrophilicity parameter, and indicate that theory can predict excited state properties of indole derivatives with electron-withdrawing substituents.
Facile heterolytic bond splitting of molecular chlorine upon reactions with Lewis bases: Comparison with ICl and I2
- First Published: 23 September 2024
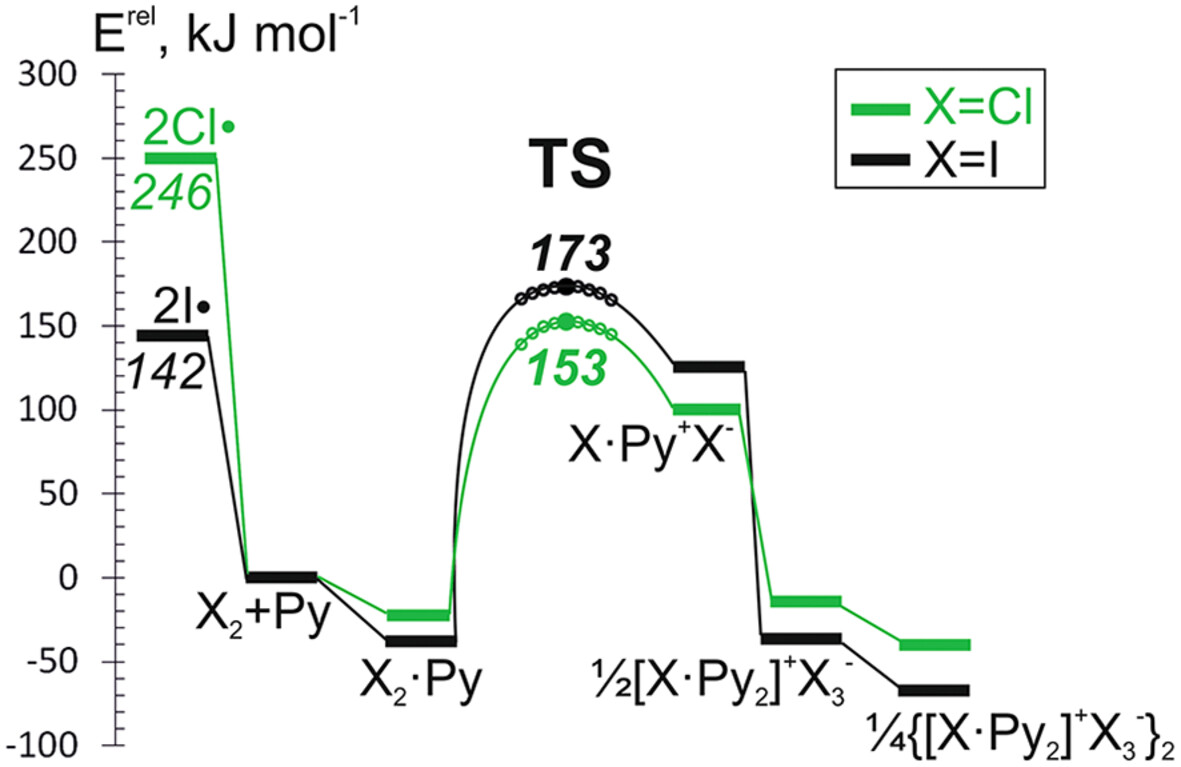
Molecular and ionic isomers of complexes in Cl2-LB systems (LB = nitrogen-containing Lewis base) were explored at M06-2X/def2-TZVPD level of theory, verified against benchmark computations at CCSD(T) ab initio level for selected compounds. Formation of contact ion pairs [Cl∙(LB)2]+…C makes the heterogeneous halogen-halogen bond splitting both energetically and kinetically favorable.
Development of a machine learning-based target-specific scoring function for structure-based binding affinity prediction for human dihydroorotate dehydrogenase inhibitors
- First Published: 26 September 2024
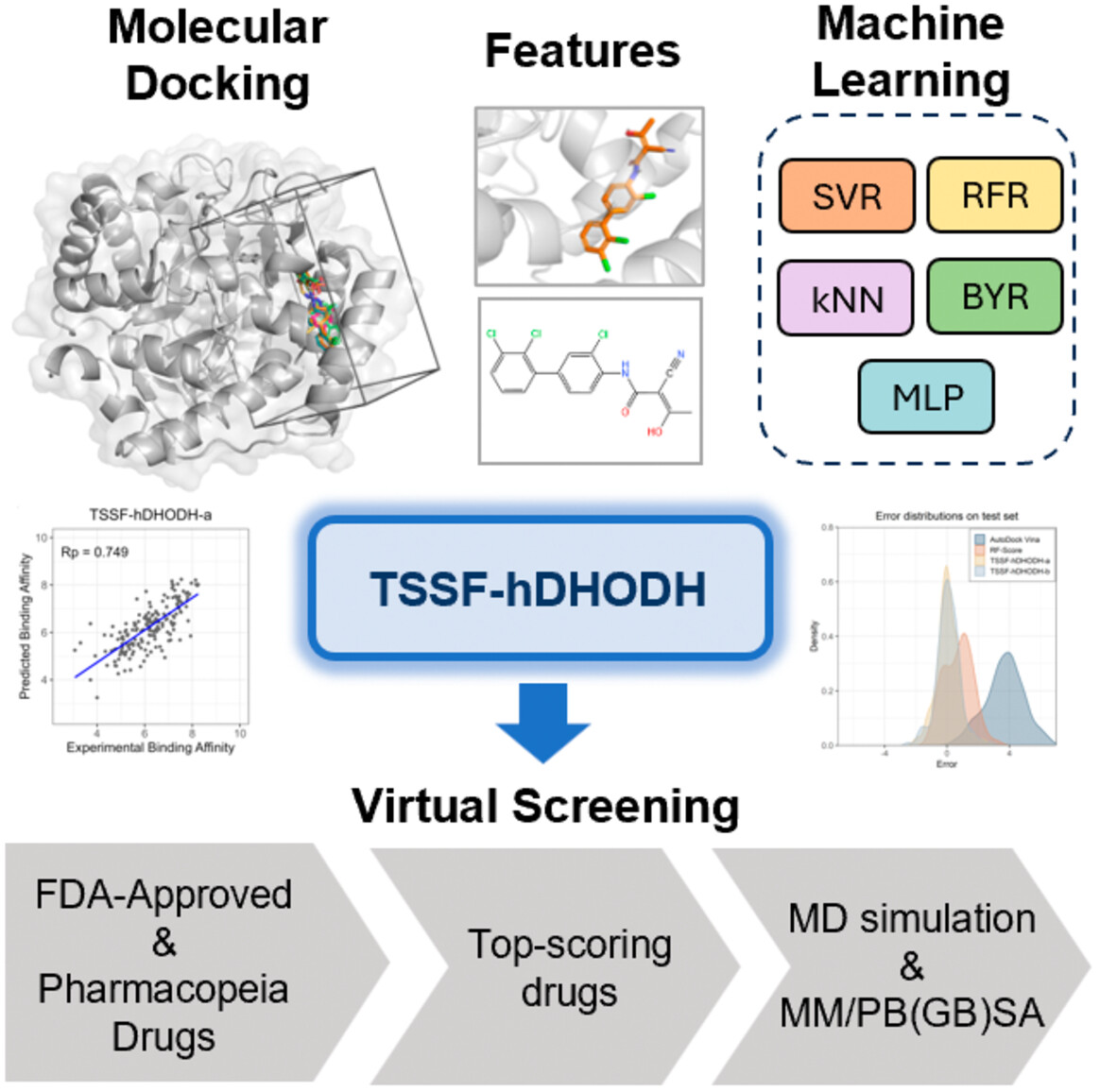
Target-specific scoring function for human dihydroorotate dehydrogenase (TSSF-hDHODH) is a machine learning-based target-specific scoring function based on molecular docking structures for predicting the binding affinities of inhibitors to hDHODH. This high-performing scoring function is developed by optimally combining interaction and ligand features with five machine learning algorithms and can be applied to virtual screening of potential inhibitors from compound libraries. This study can aid in the development of drugs for hDHODH and scoring functions for other targets.
Theoretical study on the luminescent and reaction mechanism of dansyl-based fluorescence probe for detecting hydrogen sulfide
- First Published: 26 September 2024
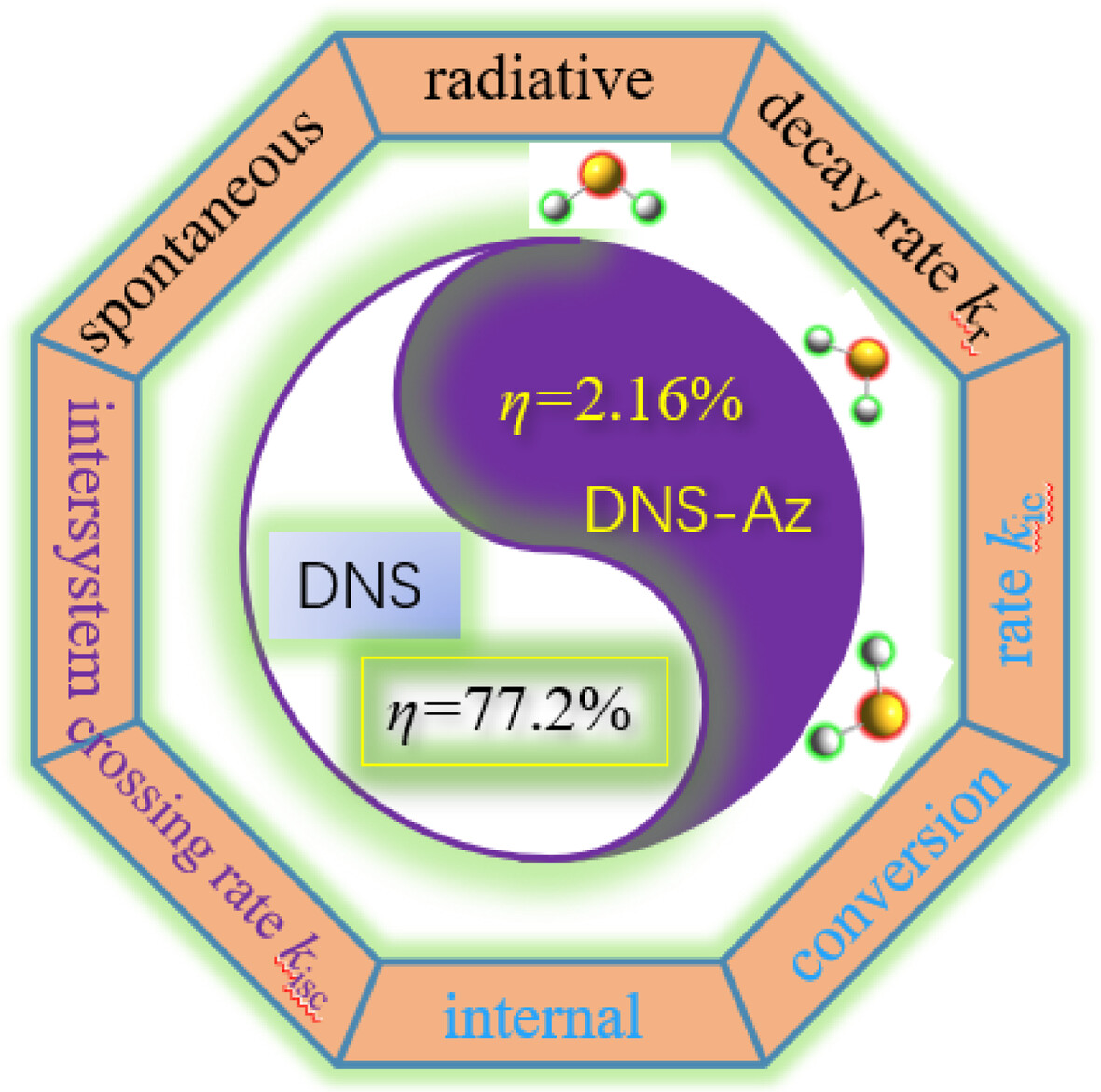
An theoretical study on the luminescent and reaction mechanism of dansyl-based fluorescence probe for detecting hydrogen sulfide is conducted and the results can explain the “turn-on” effect of fluorescent probe in the presence of H2S.
Hydride and halide abstraction reactions behind the enhanced basicity of Be and Mg clusters with nitrogen bases
- First Published: 28 September 2024
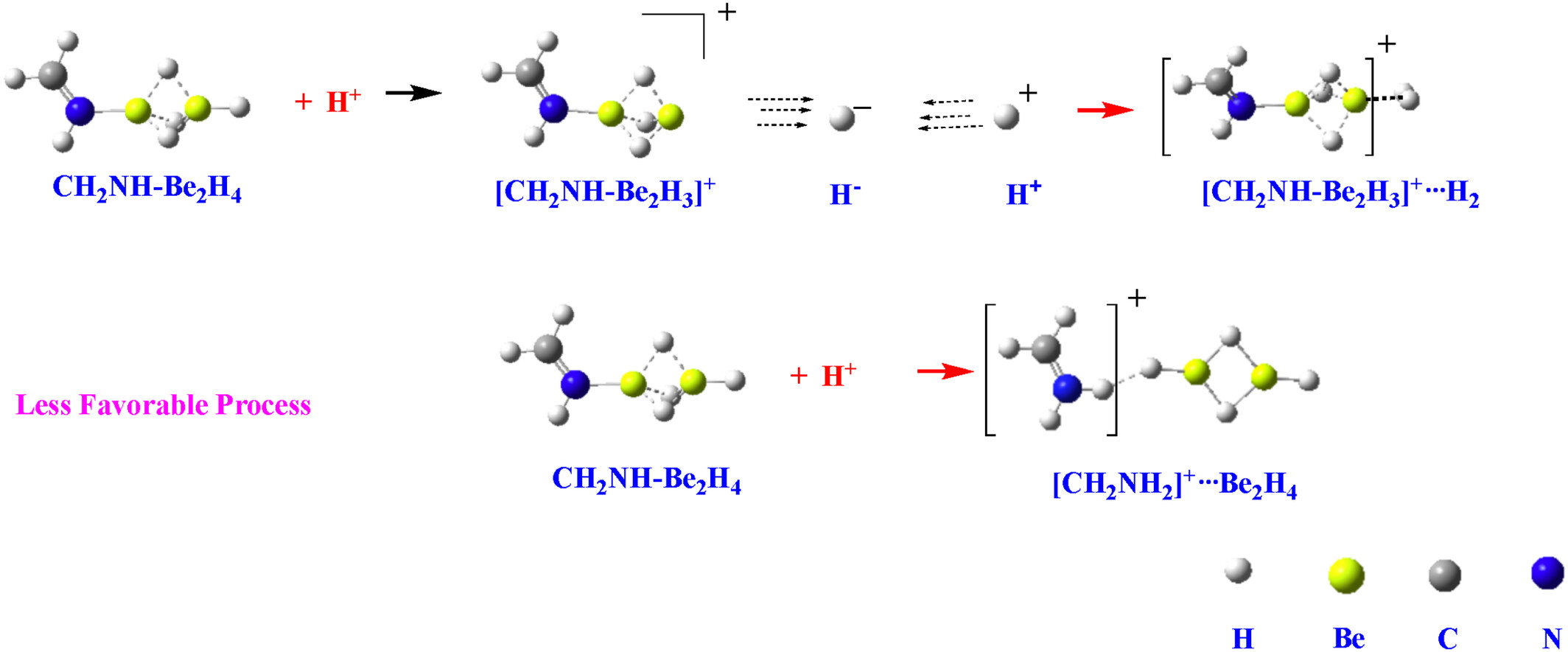
In the protonation of MX2 and M2X2 (M = Be, Mg; X = H, H) complexes with nitrogen bases, the hydrogen or fluoride abstraction process competes with the protonation at the base. The abstraction process is consistently favored for hydrides.
Fluxional halogen bonds in linear complexes of tetrafluorodiiodobenzene with dinitrobenzene
- First Published: 01 October 2024
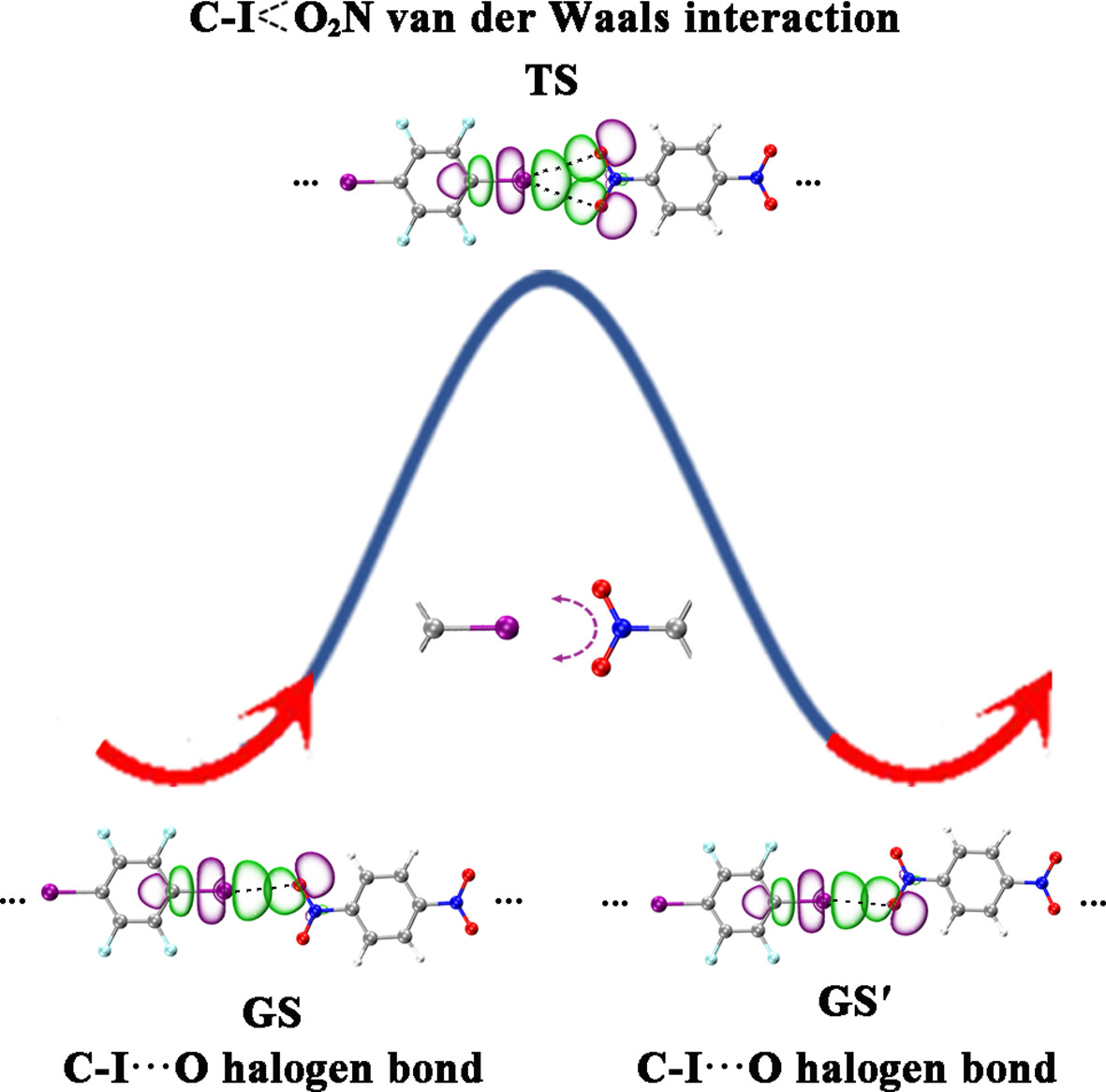
Detailed noncovalent bonding analyses unveil the existence of a fluxional halogen bonds (FXBs) in a series of linear (IC6F4I)m(OONC6H4NOO)n (m + n = 5) complexes which fluctuates reversibly in a linear mechanism, facilitating the fluxionality of the systems.
Theoretical design of new ligands to boost reaction rate and selectivity in palladium-catalyzed aromatic fluorination
- First Published: 01 October 2024
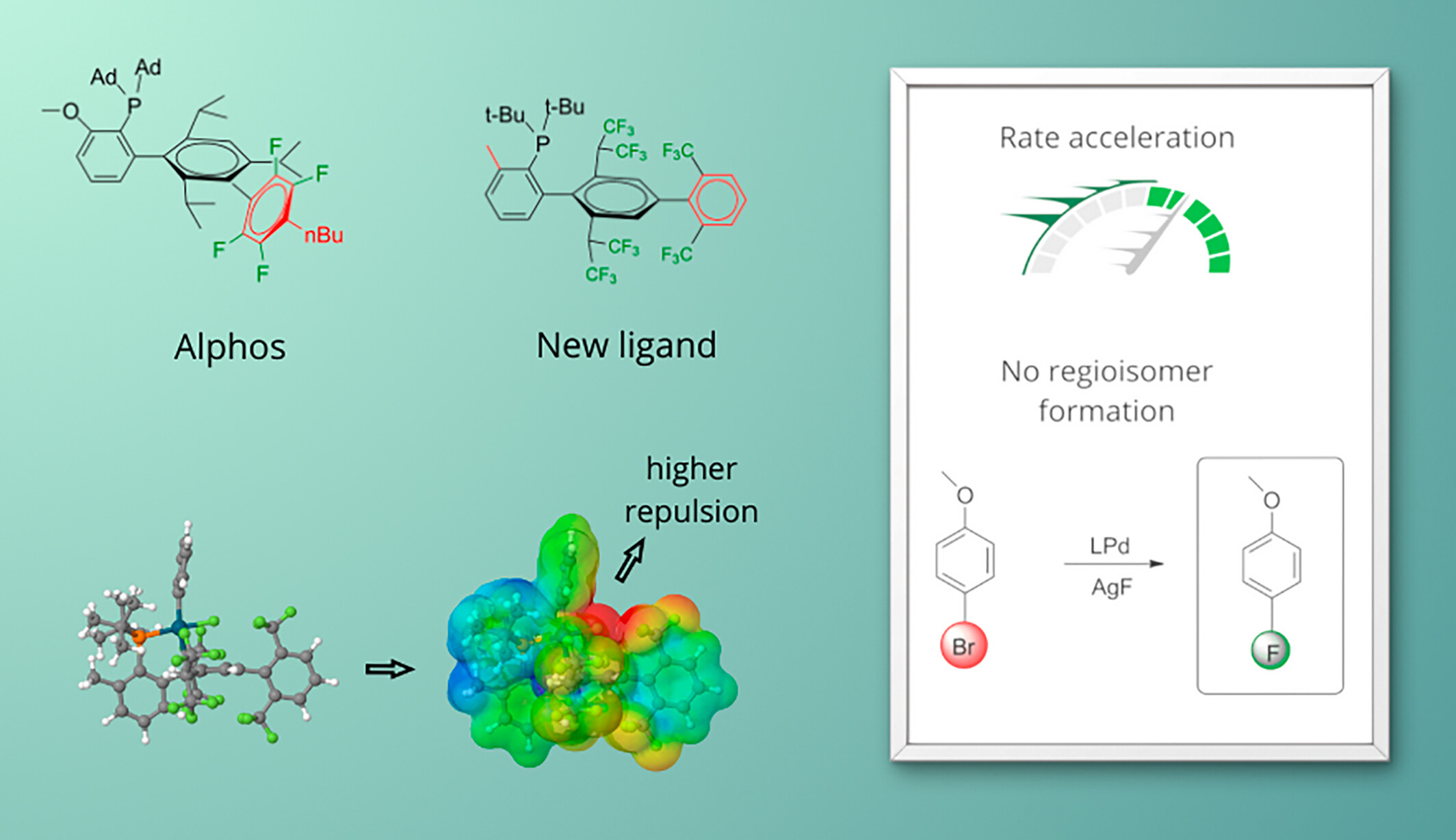
A new biaryl monophosphine ligand designed by theoretical calculations with negative electrostatic potential close to the palladium-bonded fluoride ion is predicted to increase the reaction rate and regioselectivity in palladium-catalyzed aromatic fluorination.
An ANI-2 enabled open-source protocol to estimate ligand strain after docking
- First Published: 05 October 2024
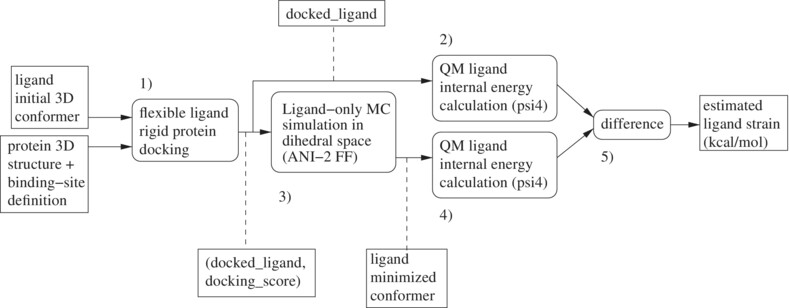
Open-source protocol to estimate ligand strain after docking: two quantum mechanics (QM) single-point energy calculations with a Monte Carlo (MC) based ligand minimization in-between. The MC simulation uses the ANI-2x (QM approximating) force field and is performed in the dihedral space.
High-throughput molecular simulations of SARS-CoV-2 receptor binding domain mutants quantify correlations between dynamic fluctuations and protein expression
- First Published: 15 October 2024
Probing the performance of DFT in the structural characterization of [FeFe] hydrogenase models
- First Published: 17 October 2024
![Probing the performance of DFT in the structural characterization of [FeFe] hydrogenase models](/cms/asset/15f69ded-f972-40b7-bf61-05d290dd119d/jcc27515-toc-0001-m.jpg)
Due to their potential for eco-friendly energetic applications, [FeFe] hydrogenase models attract interest of both experimental and computational chemists. Here, a series of DFT methods was tested to establish their performance in describing the molecular and crystal structures of recently synthesized [FeFe] hydrogenase models. r2SCAN is recommended for predicting the structure of such models with a high degree of accuracy.
Fragment and torsion biasing algorithms for construction of small organic molecules in proteins using DOCK
- First Published: 22 October 2024
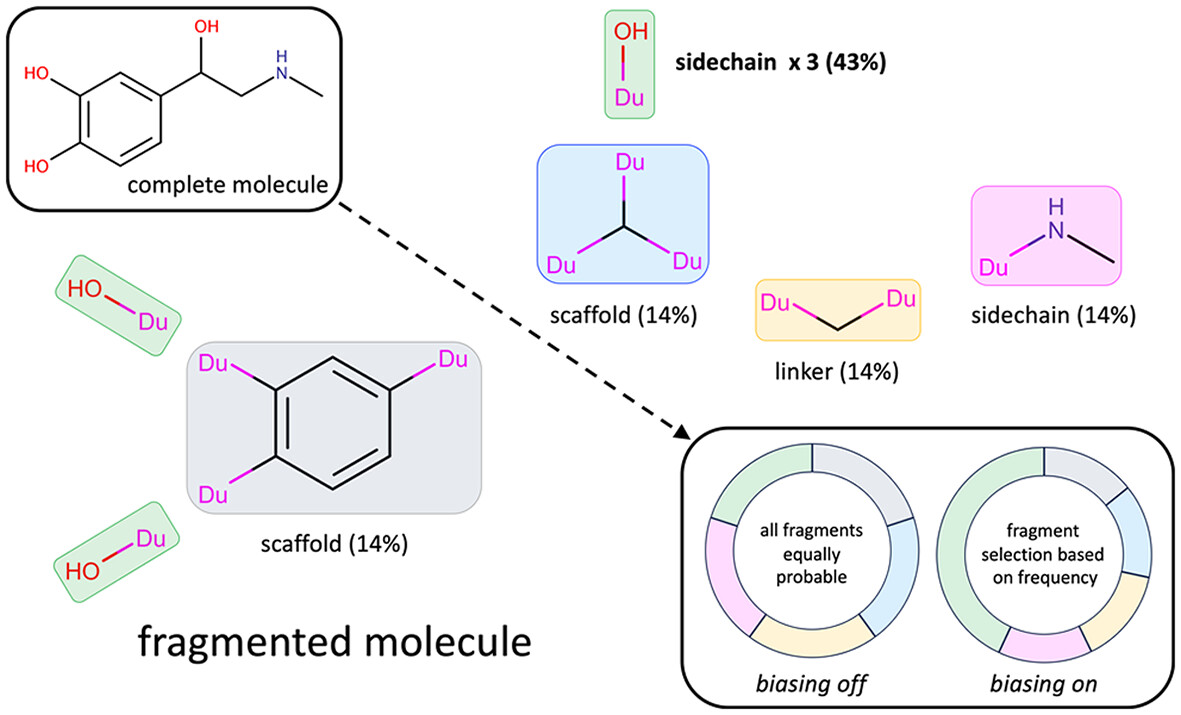
Fragment-based de novo design can construct new molecules from scratch from an input library of allowable fragments and torsion types. In this work, a new series of biasing methods have been developed for the program DOCK6, which facilitate the construction of molecular ensembles containing fragment and torsion populations in proportion to those seen in drug-like libraries. Changes to clustering and pruning have also been made to increase the overall efficiency of the methods, producing many more unique molecules in a similar amount of time.
Pre-training strategy for antiviral drug screening with low-data graph neural network: A case study in HIV-1 K103N reverse transcriptase
- First Published: 22 October 2024
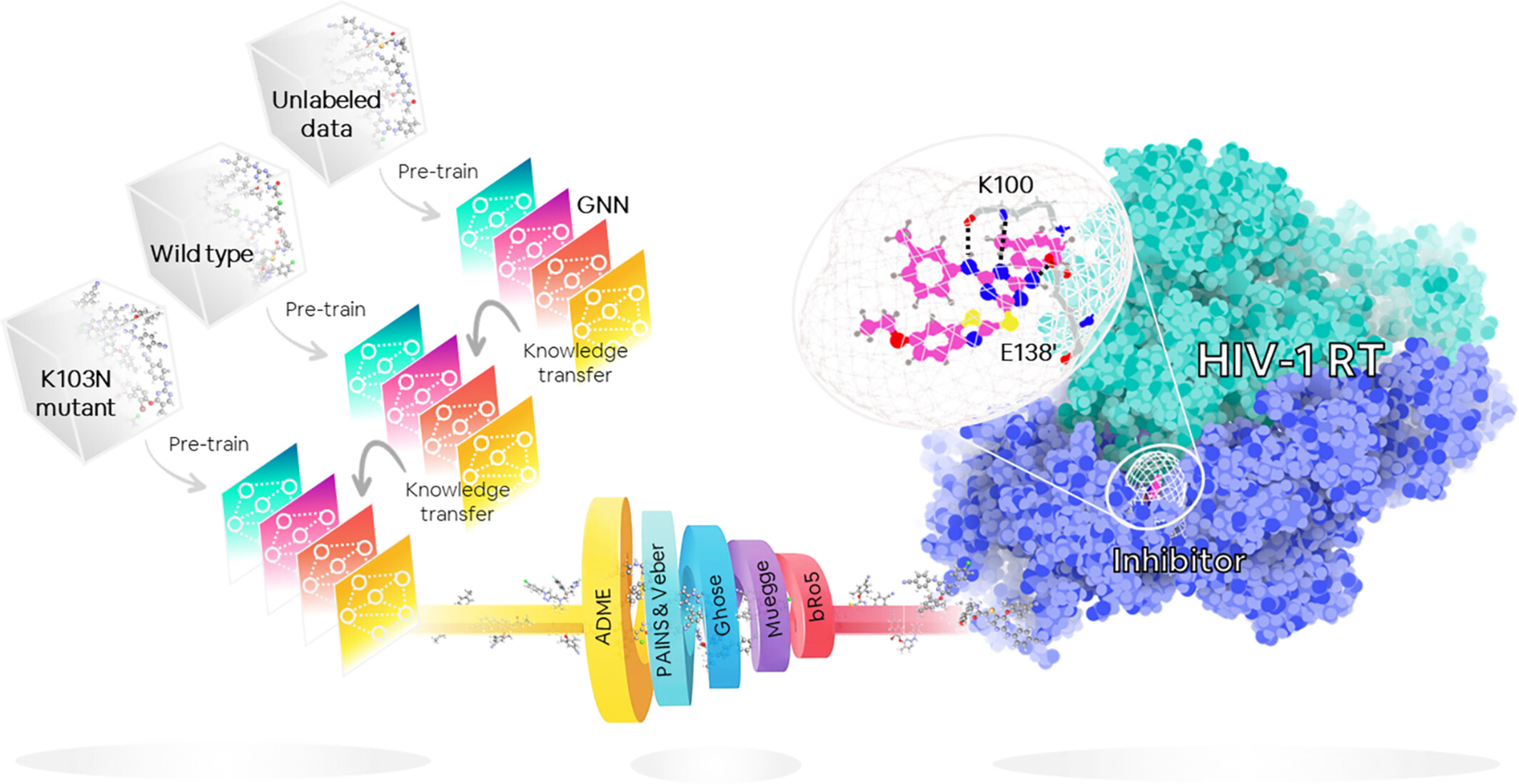
Self-supervised pre-training pipeline integrated with high-throughput virtual screening for non-nucleoside reverse transcriptase inhibitors (NNRTIs).
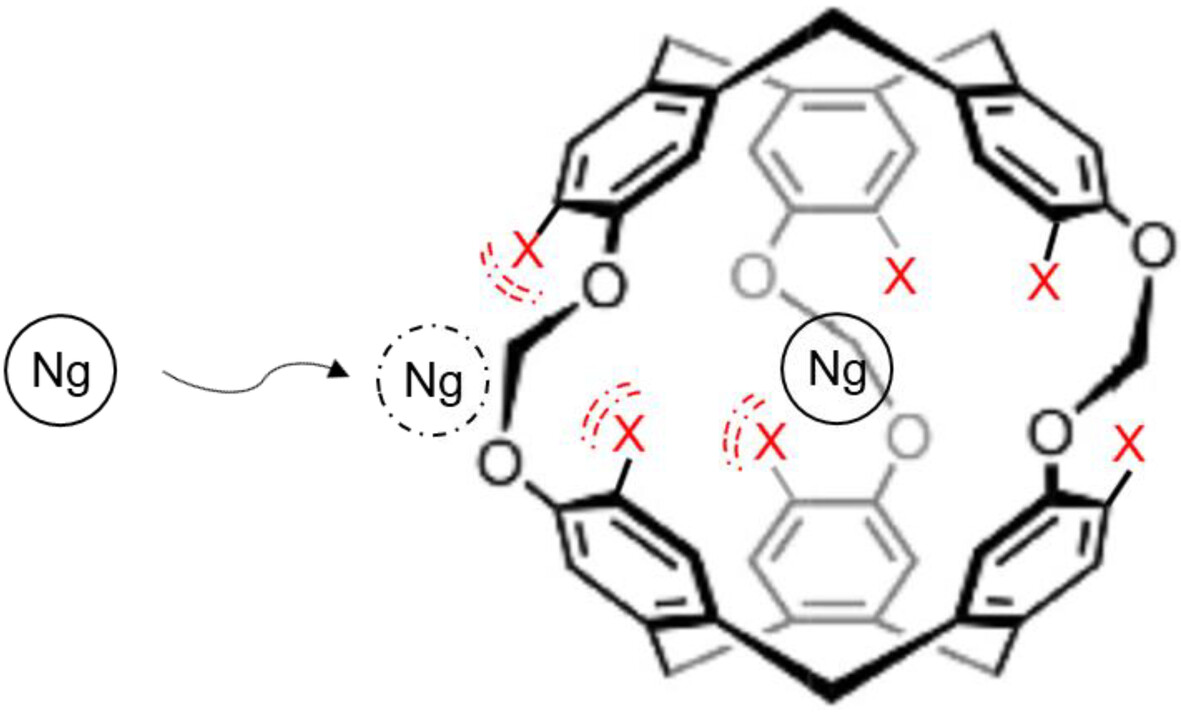
Investigation of the complete encapsulation process of the noble gases by cryptophanes
- First Published: 24 October 2024
Assessing small molecule conformational sampling methods in molecular docking
- First Published: 30 October 2024
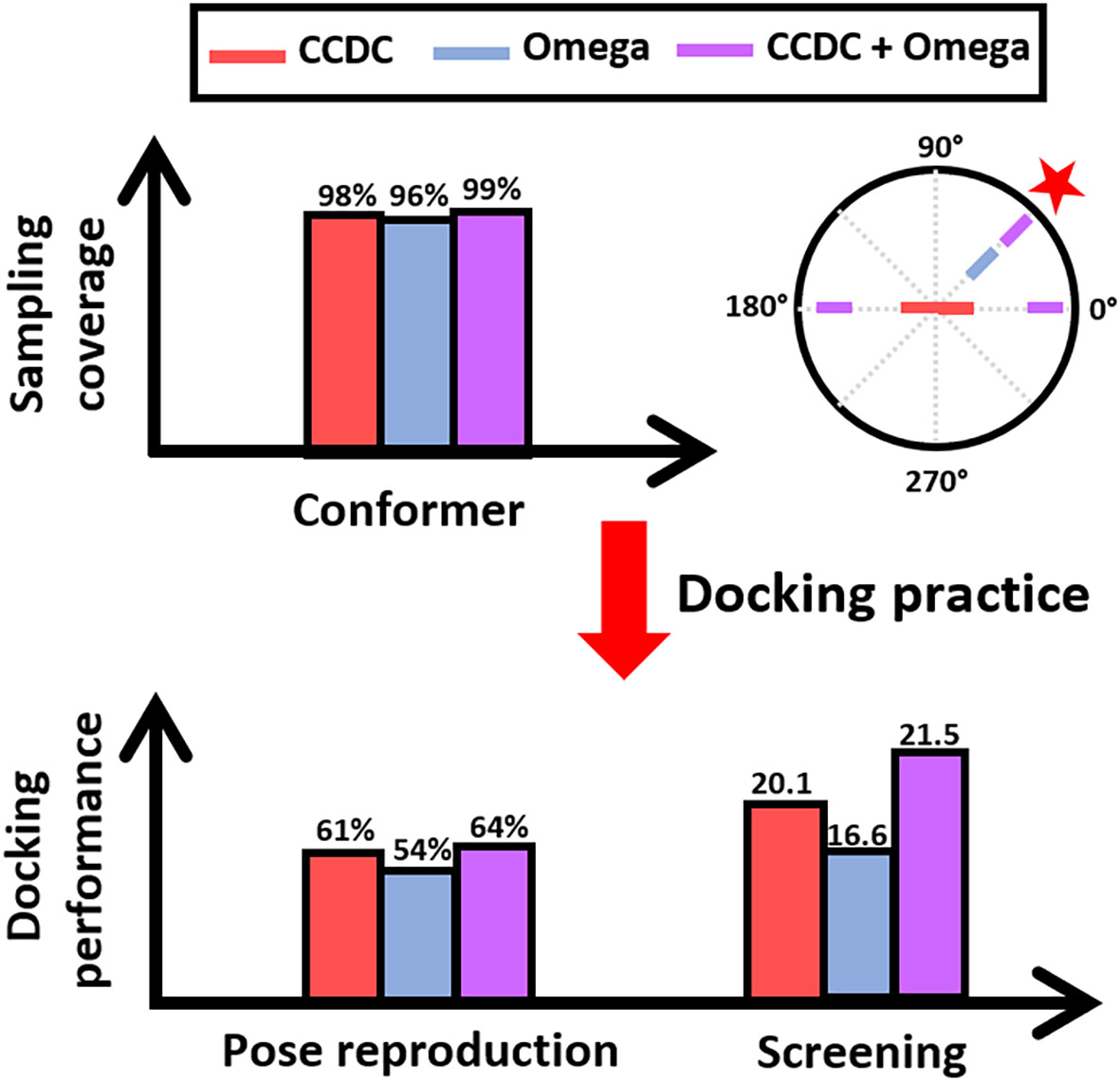
Small molecule conformational sampling is crucial for exploring the vast conformational space within specific chemical environments. Recent advancements have led to the emergence of various conformational sampling methods. This study evaluates the impact of different sampling methods on molecular docking. Among the methods, the CCDC Conformer Generator demonstrates superior bioactive pose reproducibility and comparable screening. Additionally, each sampling method excels in specific cases, and combining various methods can enhance the overall performance.
On delivering polar solvation free energy of proteins from energy minimized structures using a regularized super-Gaussian Poisson–Boltzmann model
- First Published: 30 October 2024
Stable, aromatic, and electrophilic azepinium ions: Design using quantum chemical methods
- First Published: 30 October 2024
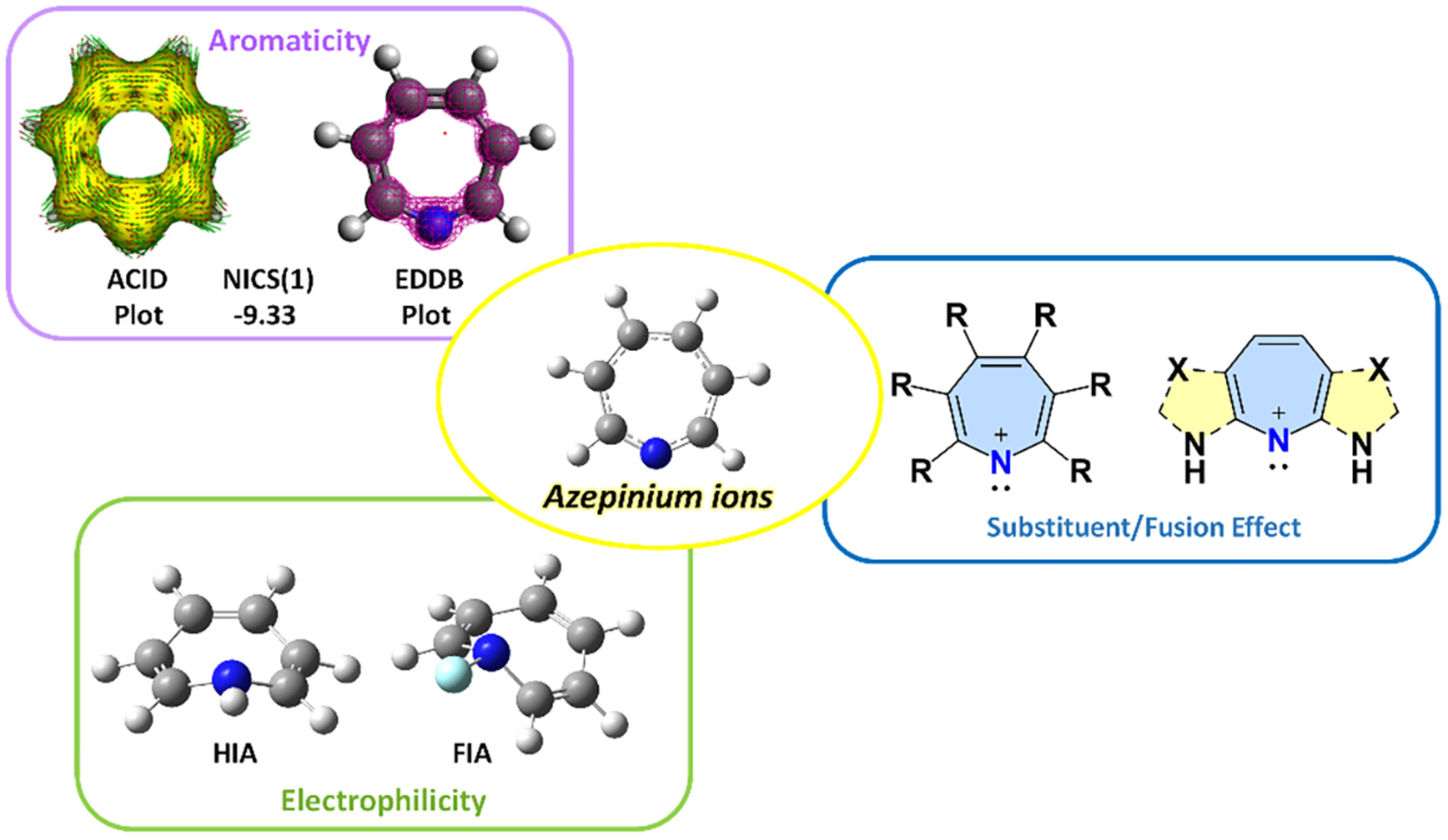
The cyclic nitrenium ions with seven membered rings are aromatic but not very stable. Their electronic, energetic properties can be fine-tuned and the species carrying ΔES-T ≈ 50 kcal/mol may prove to be stable.
Excited state relaxation mechanisms of paracetamol and acetanilide
- First Published: 04 November 2024
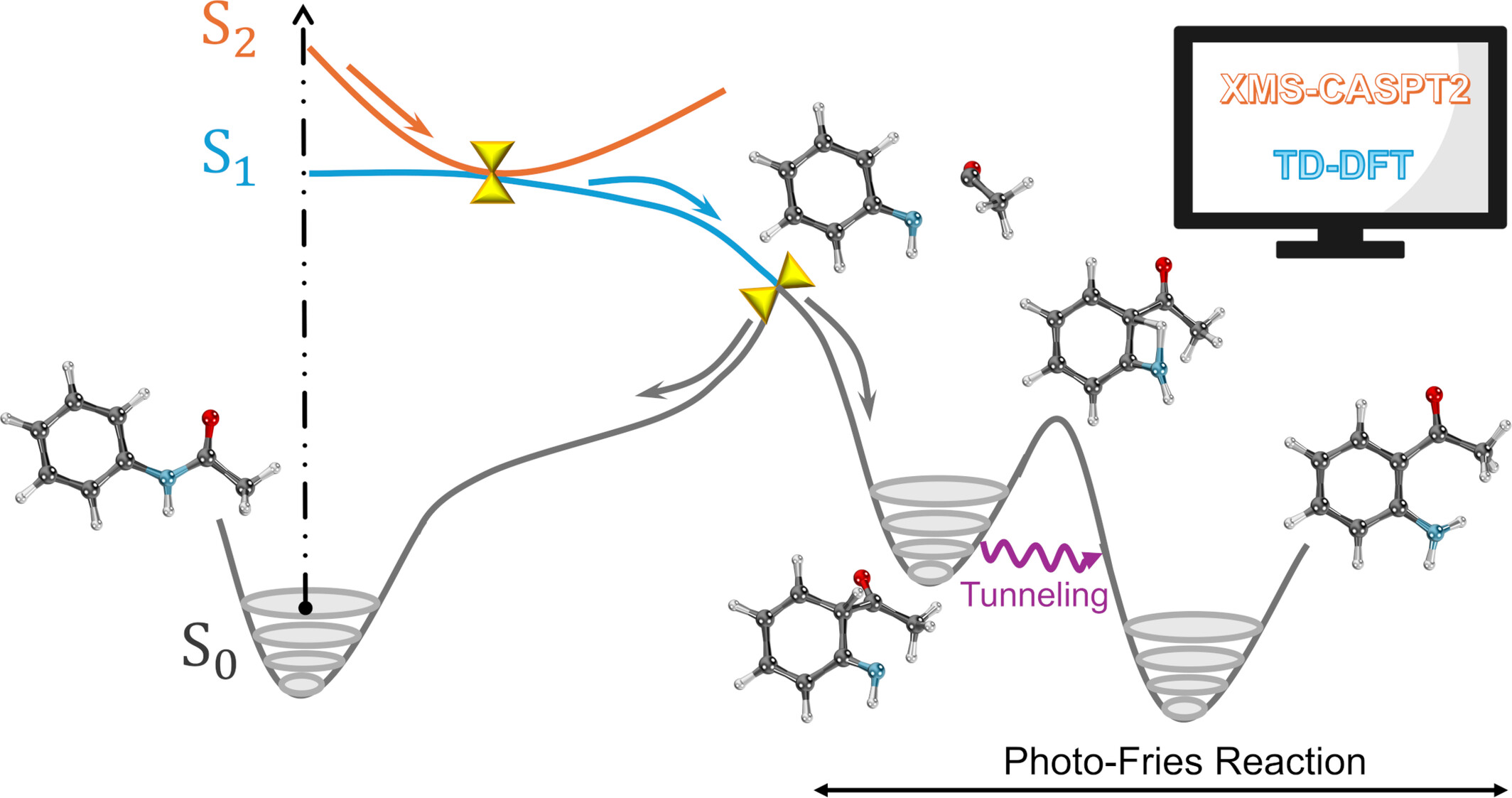
Acetanilide and paracetamol photophysics after the photoexcitation to the second singlet excited state, which can lead to the formation of the photo-fries photoproduct.
DC24: A new density coherence functional for multiconfiguration density-coherence functional theory
- First Published: 08 November 2024
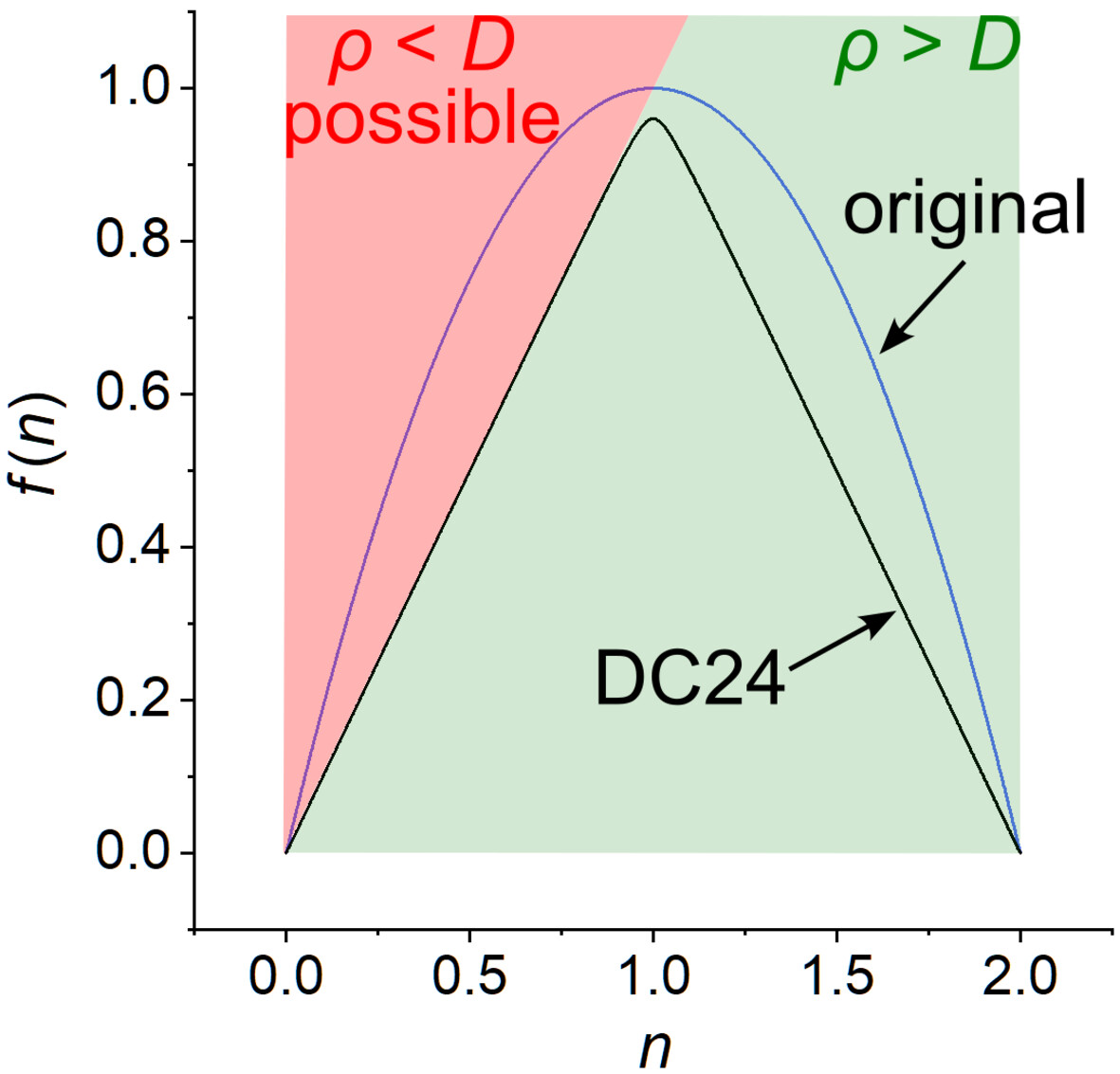
Multiconfiguration density-coherence functional theory is a low-cost and high-accuracy electronic structure method that recovers dynamic correlation in inherently multiconfigurational molecules and transition states by expressing the nonclassical energy as a functional of the unpaired density computed from the one-particle reduced density matrix in the coordinate representation. Here we propose a new, more physical expression for the unpaired density and use it to optimize a density coherence functional with improved physical interpretation and upgraded accuracy.
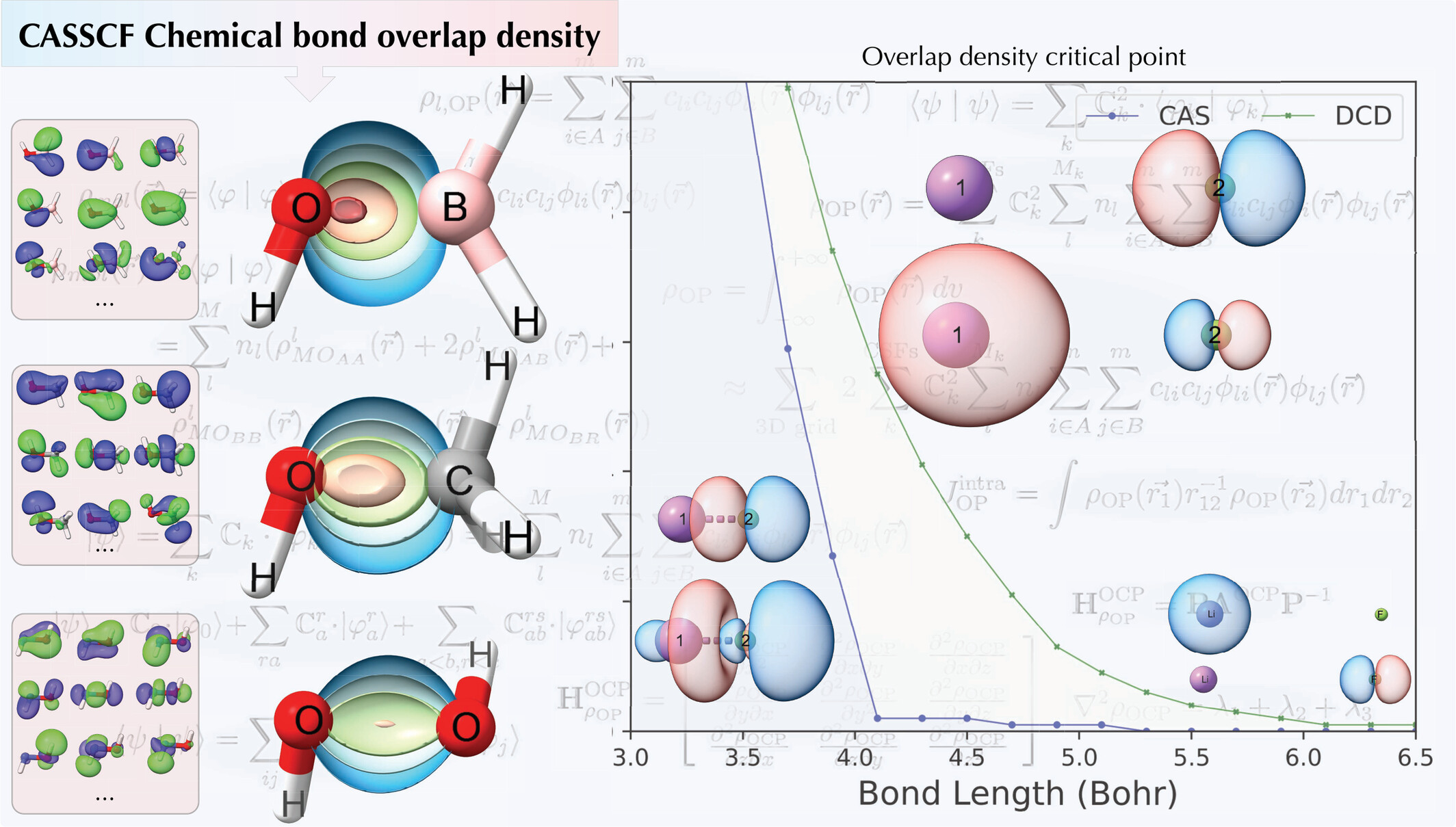
Chemical Bond Overlap Descriptors From Multiconfiguration Wavefunctions
- First Published: 28 November 2024
Unveiling the Antiferromagnetic Properties of Cr2Pbn (n = 3–20) Clusters
- First Published: 30 November 2024
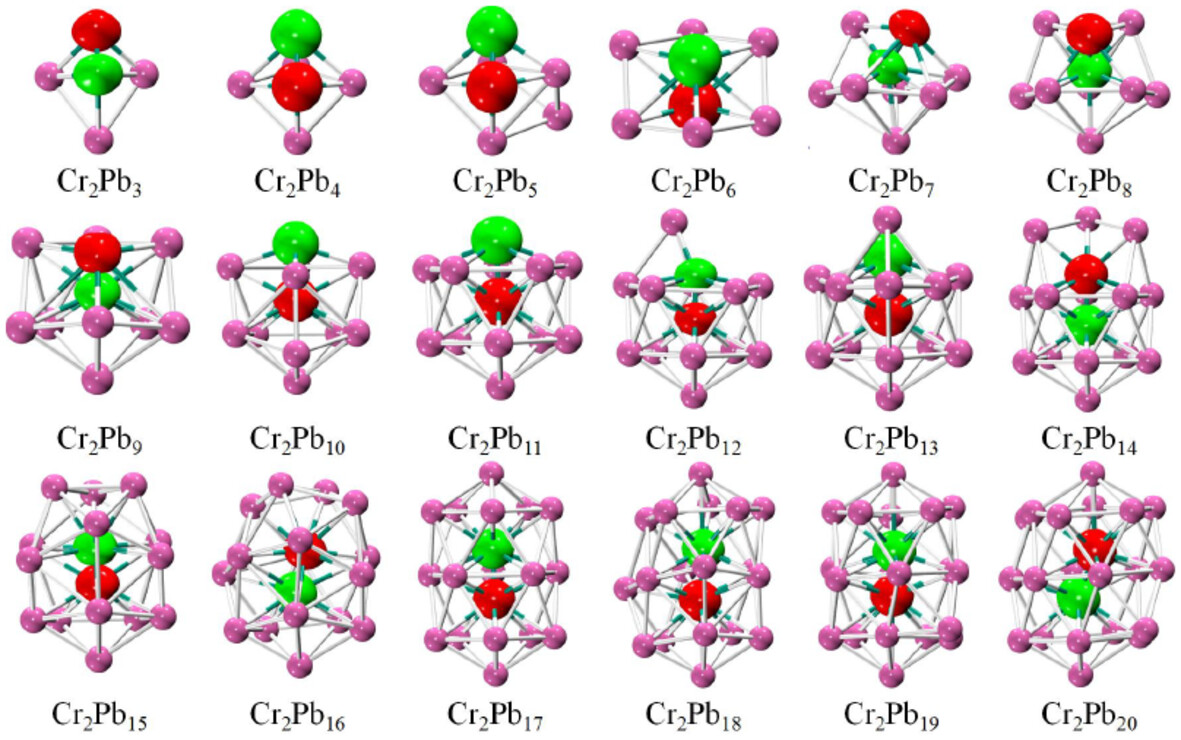
In this work, we investigated the structures and electronic and magnetic properties of Cr2Pbn (n = 3–20) clusters based on a self-developed genetic algorithm coupled with density functional theory calculations. It found that all Cr2Pbn clusters are antiferromagnets, except for the ferrimagnetic Cr2Pb11 with a net magnetic moment of 2 μB. The discovered stable Cr2Pb17 cluster can assemble into dimers and trimers while maintaining its geometric structure and antiferromagnetic properties, indicating the potential of becoming a structural unit for cluster-assembled antiferromagnetic materials.
SOFTWARE NOTE
MolAR: Memory-Safe Library for Analysis of MD Simulations Written in Rust
- First Published: 01 December 2024
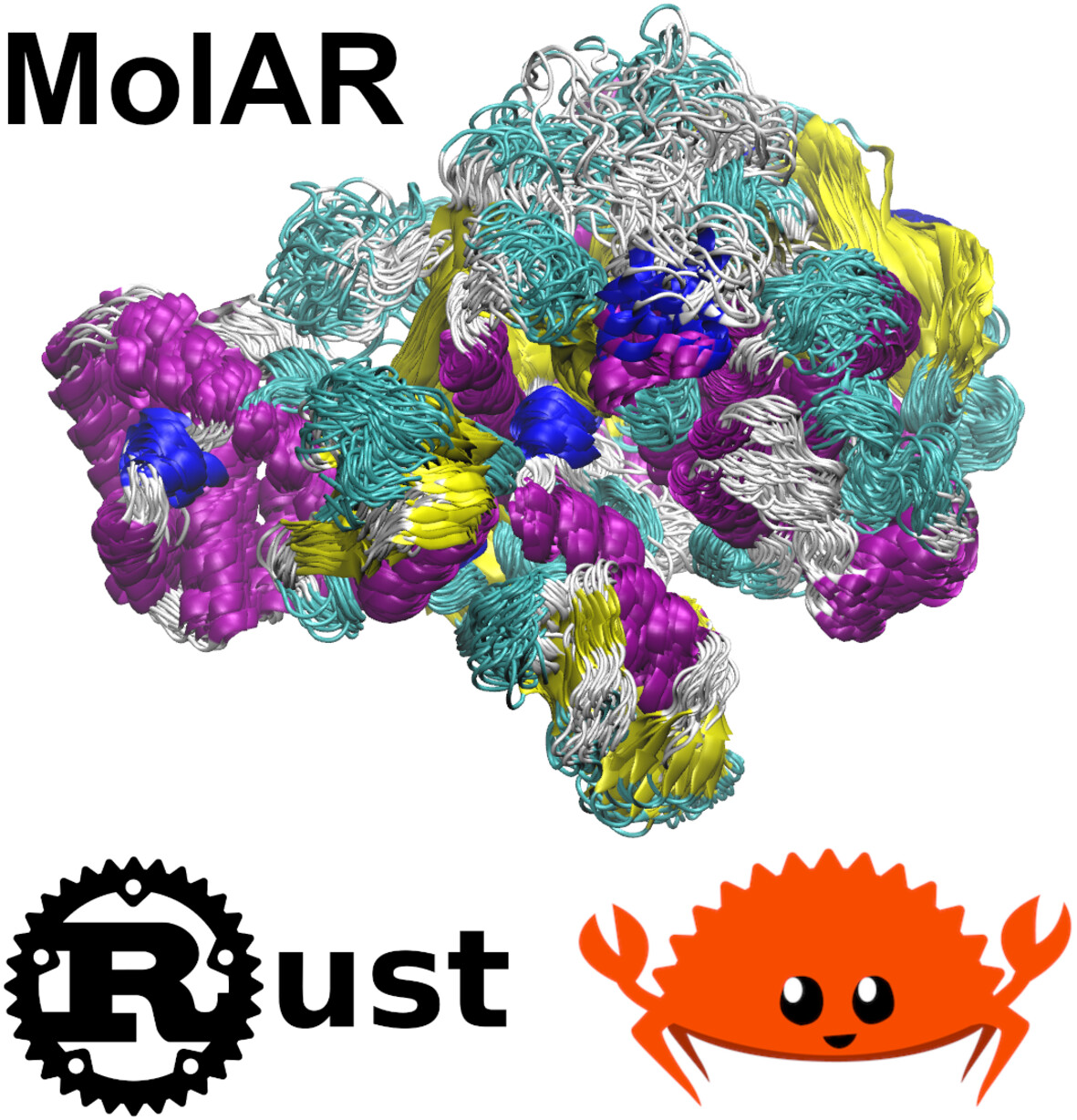
MolAR is the first memory-safe library for analysis of MD simulations written in Rust. MolAR is intended to explore the advantages and challenges of implementing molecular analysis software in the memory-safe natively compiled language and to develop specific memory-safe abstractions for this kind of software. MolAR outperforms popular molecular analysis libraries and tools, which makes it attractive for implementing computationally intensive analysis tasks.
RESEARCH ARTICLE
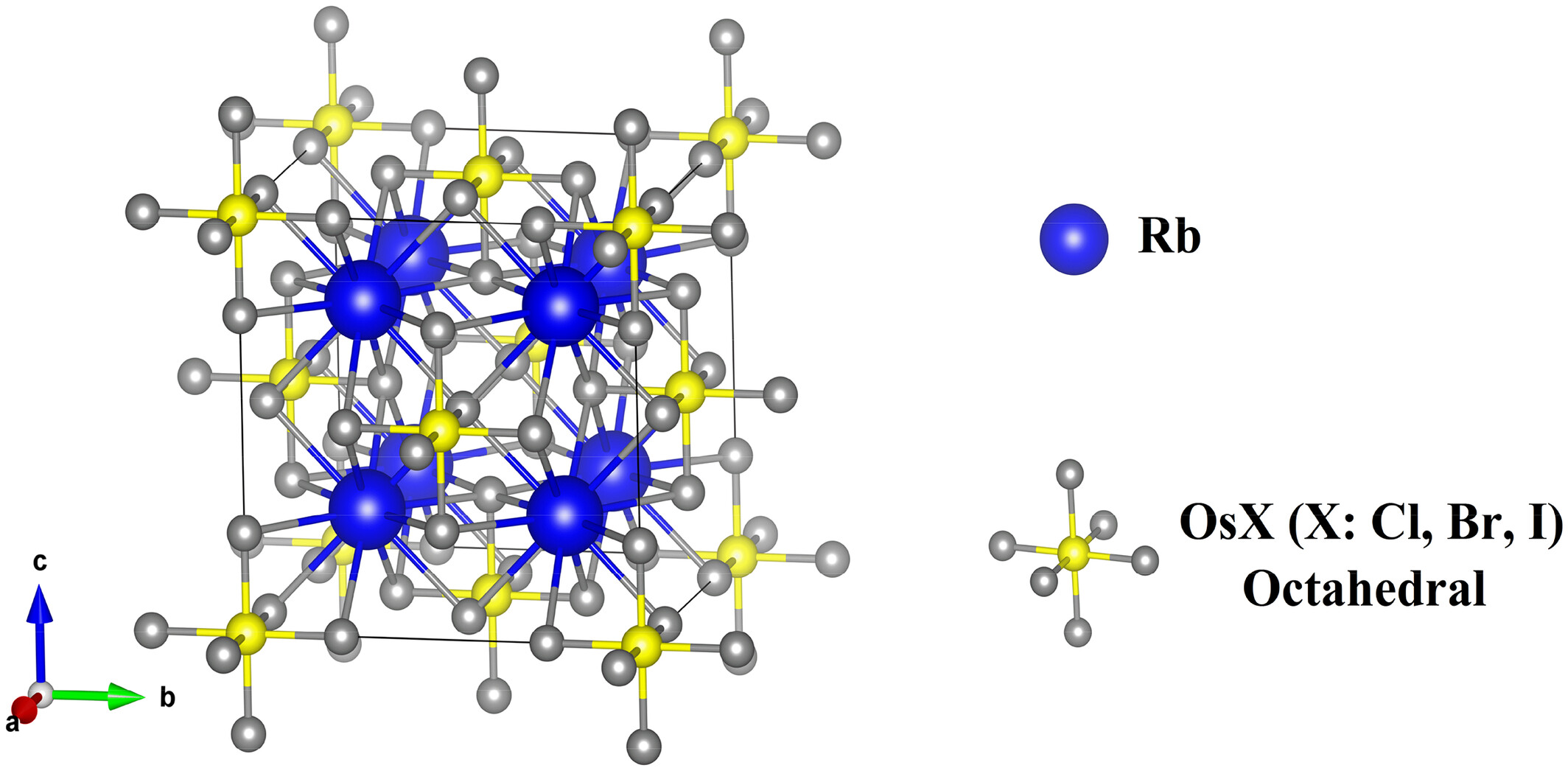
A First-Principle Study Investigating the Half-Metallic and Mechanical Properties of Double Halide Perovskites Rb2OsX6 (X = cl, Br, and I) for Spintronic Applications
- First Published: 05 December 2024
Assessment of DFT Functionals for Predicting the Magnetic Exchange Coupling Constants of Nonalternant Hydrocarbon Diradicals: The Role of Hartree–Fock Exchange
- First Published: 05 December 2024
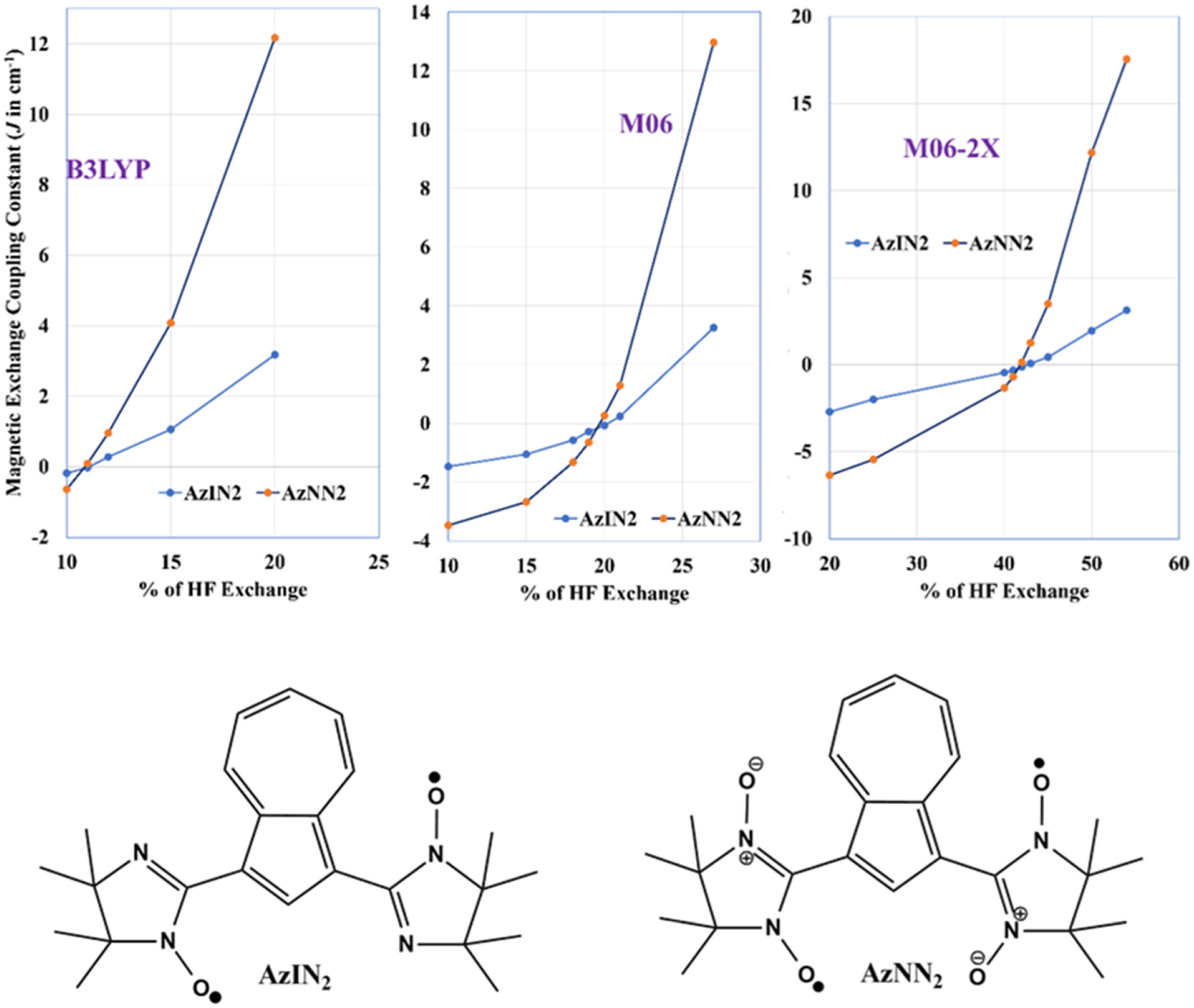
The magnetic exchange coupling constant calculation for azulene-bridged diradicals is highly sensitive to the amount of Hartree–Fock (HF) exchange in the density functionals. The hybrid functionals accurately determine the sign of ferromagnetic coupling. In a particular value of HF exchange in the hybrid functionals, they predict the ferro- and antiferromagnetic coupling correctly.
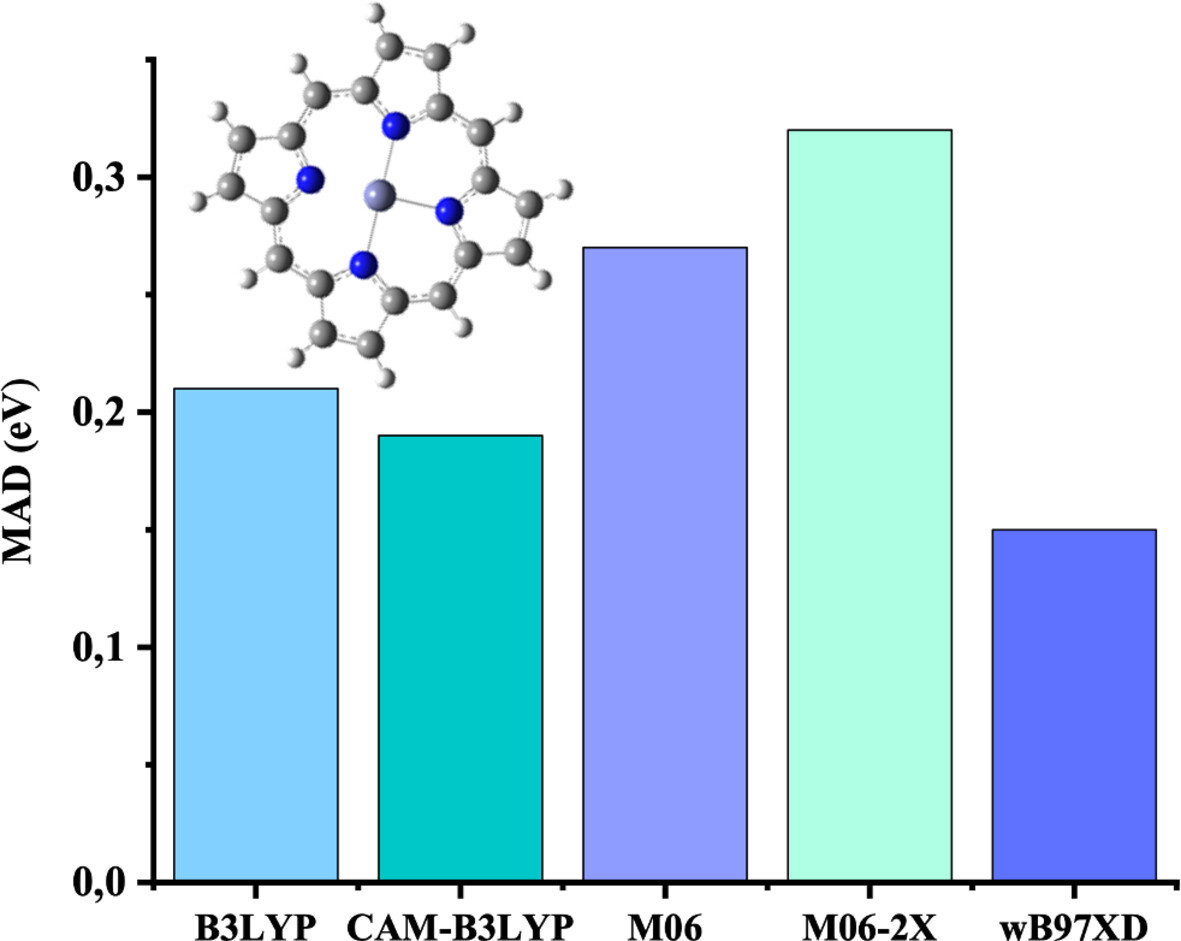
Density Functional Theory (DFT) and Time-Dependent DFT (TDDFT) Studies of Porphyrin Adsorption on Graphene: Insights on the Effect of Substituents and Central Metal on Adsorption Energies
- First Published: 05 December 2024
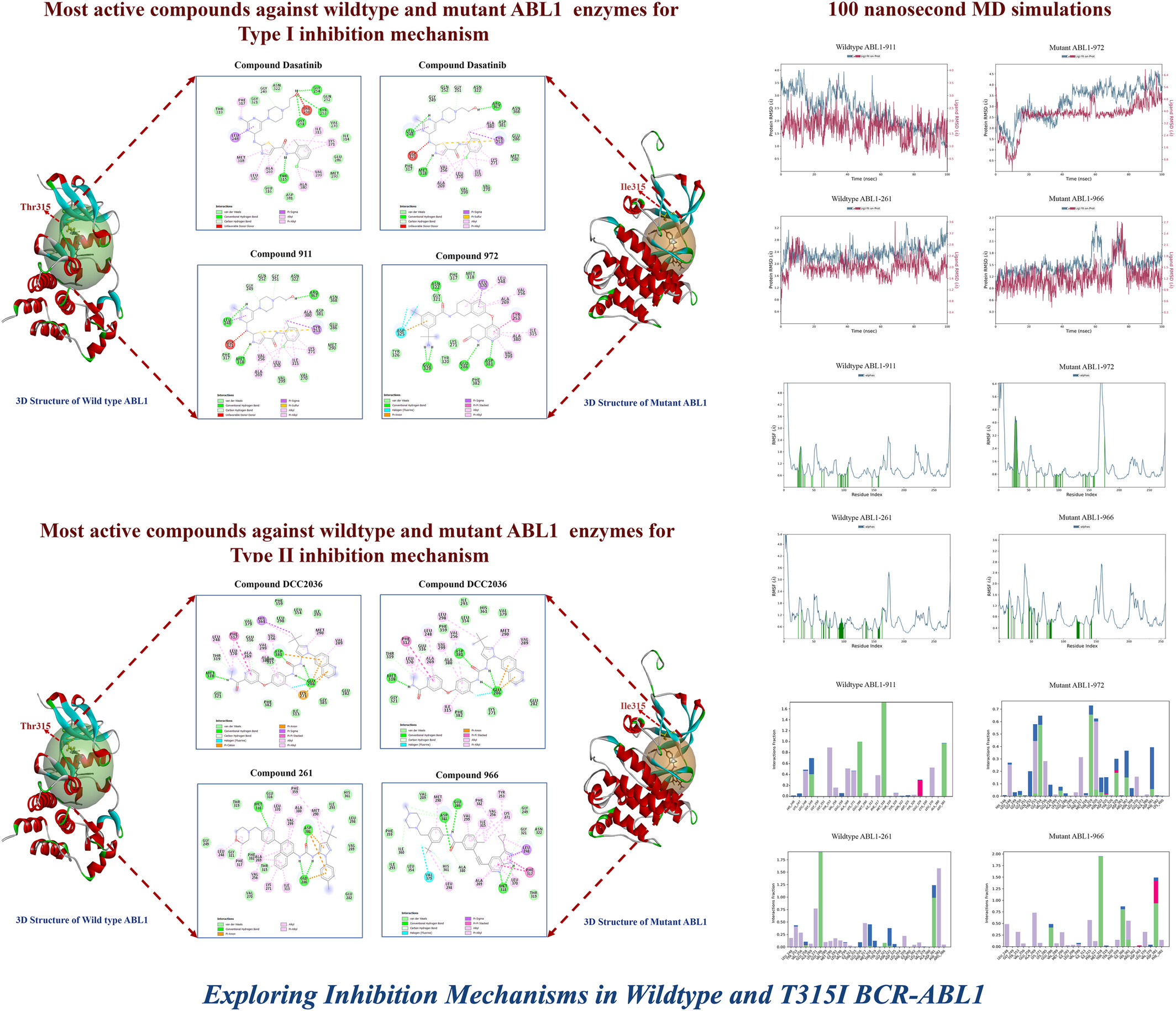
Exploring Inhibition Mechanisms in Wildtype and T315I BCR-ABL1: An In Silico Approach Integrating Virtual Screening, MD Simulations, and MM-GBSA Analysis
- First Published: 05 December 2024
Exploring the Possibility of a Planar Tetracoordinate Boron in BXY3 (X = B, Al, Ga; Y = C, Si, Ge) Clusters: A Theoretical Study
- First Published: 05 December 2024
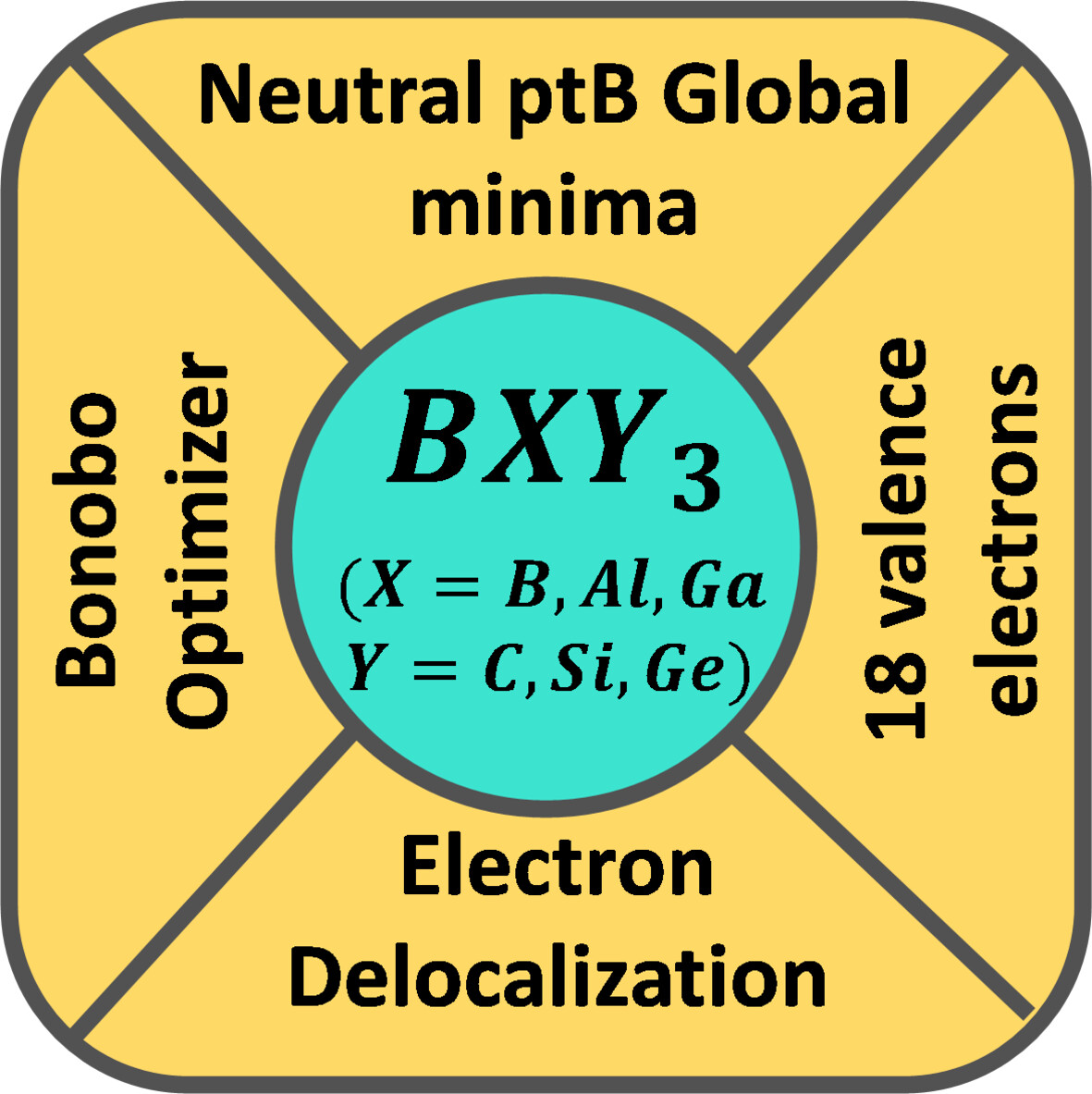
Global optimization was performed on the BXY3 (X = B, Al, Ga; Y = C, Si, Ge) clusters and neutral planar tetracoordinated boron containing global minimum structures with 18 valence electrons, were obtained for most of the systems. The ptB global minimum structures are stable at 500 K temperature and the aromaticity analysis revealed both σ- and π-electron delocalization.
Infrared Spectroscopy of Ethanethiol Monomers and Dimers at MP2 Level: Characterizing the Dimer Formation and Hydrogen Bond
- First Published: 05 December 2024
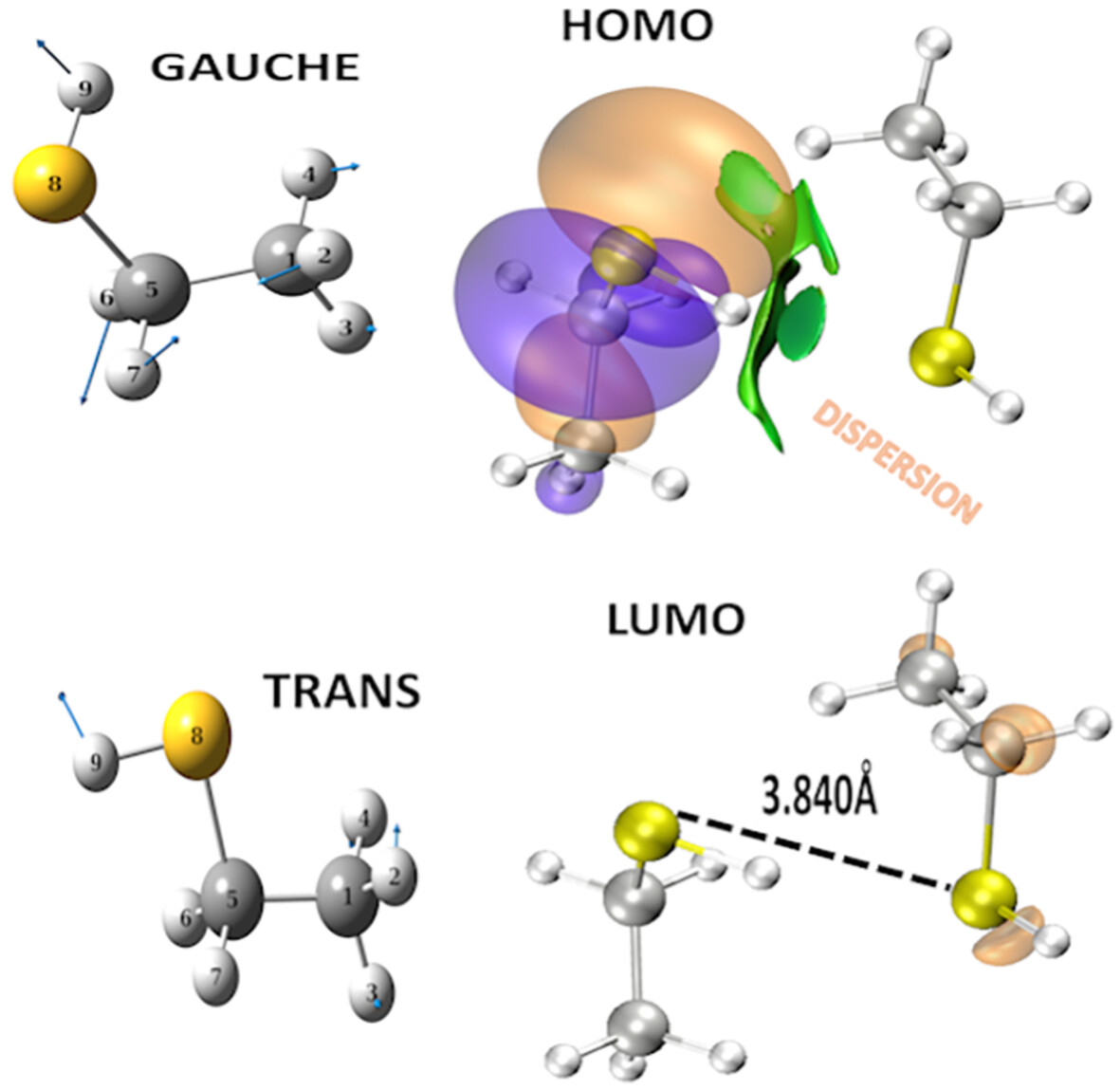
The local mode decomposition revealed that the CH2 rocking mode, associated with the CSH bending, appears only for the gauche isomer in accordance with the experimental assignments. The most stable dimer is the trans(right)–gauche(left) structure. The HOMO orbital is located on the donor monomer, and the LUMO orbital is located on the acceptor monomer, supporting the largest redshift of 33 cm−1 obtained for the gauche isomer. The configuration shows the S–H donor directed to the S acceptor with the NCI isosurface. The LUMO orbital also shows the S⋅⋅⋅S interatomic distance. Dispersion is the dominant term in this dimer, while electrostatic interactions comprise 83% of dispersion. ELF values at the critical point of S–H⋅⋅⋅S interaction describe an almost linear form of the interaction energy.
Mechanism of Ampicillin Hydrolysis by New Delhi Metallo-β-Lactamase 1: Insight From QM/MM MP2 Calculation
- First Published: 05 December 2024
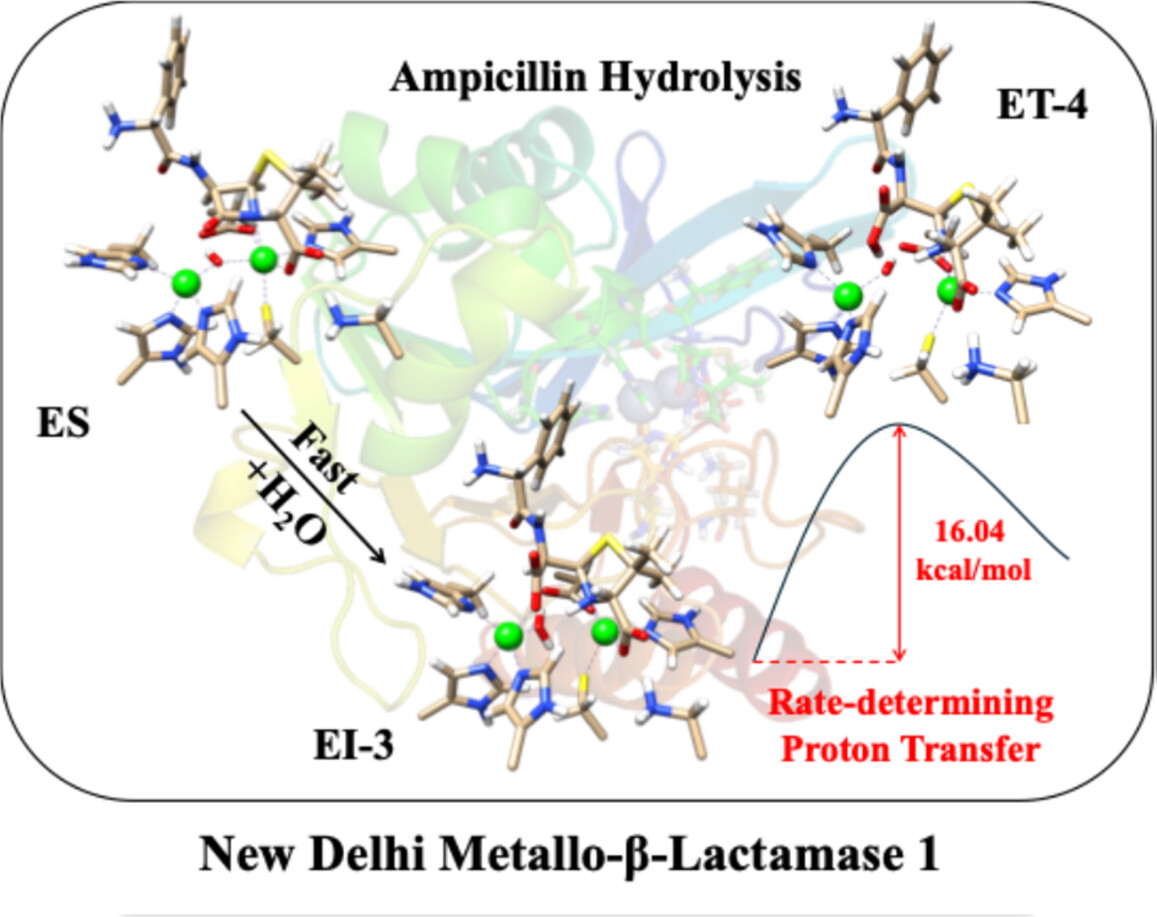
QM/MM MP2 calculations bring new insights into the catalytic mechanism of ampicillin hydrolysis by New Delhi Metallo-β-lactamase 1 (NDM-1). It is found that the rate-determining step is the dissociation of the hydrolyzed ampicillin via a solvent proton transfer to the newly form carboxylate group.
Single-Walled ZnSe Nanotubes for High-Performance Photodetectors: A Computational Prediction
- First Published: 05 December 2024
Theoretical Study of the Subsequent Decomposition Mechanisms of 1,1-Diamino-2,2-dinitroethene (FOX-7)
- First Published: 05 December 2024
Modulating the oxygen affinity of porphyrins with metals, ligands, and functional groups: A DFT study
- First Published: 06 December 2024
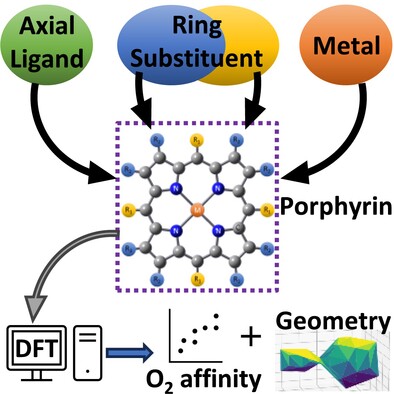
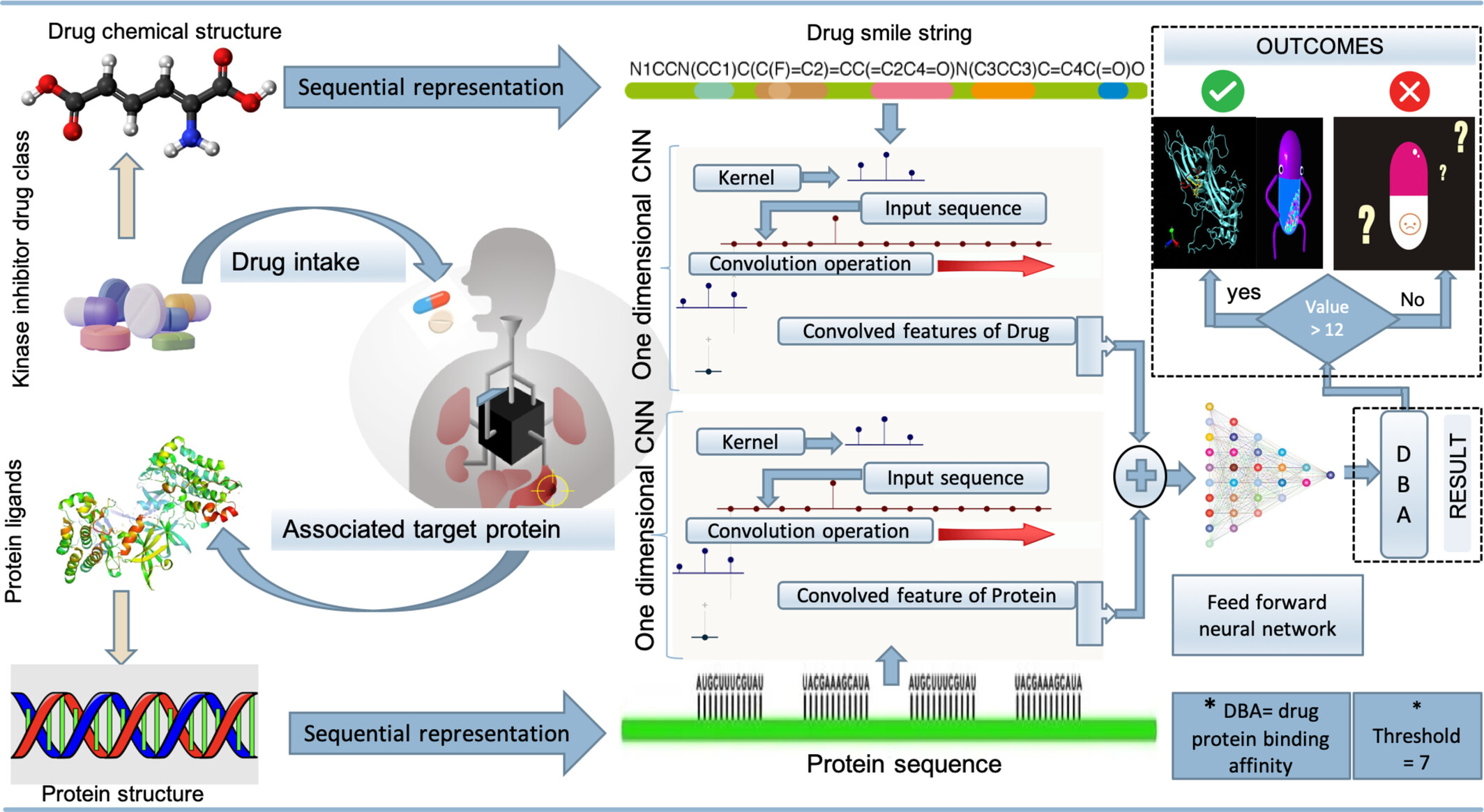
SeqDPI: A 1D-CNN approach for predicting binding affinity of kinase inhibitors
- First Published: 07 December 2024
Theoretical investigation of structure and electronic properties in Cisplatin-citrate complexes
- First Published: 07 December 2024
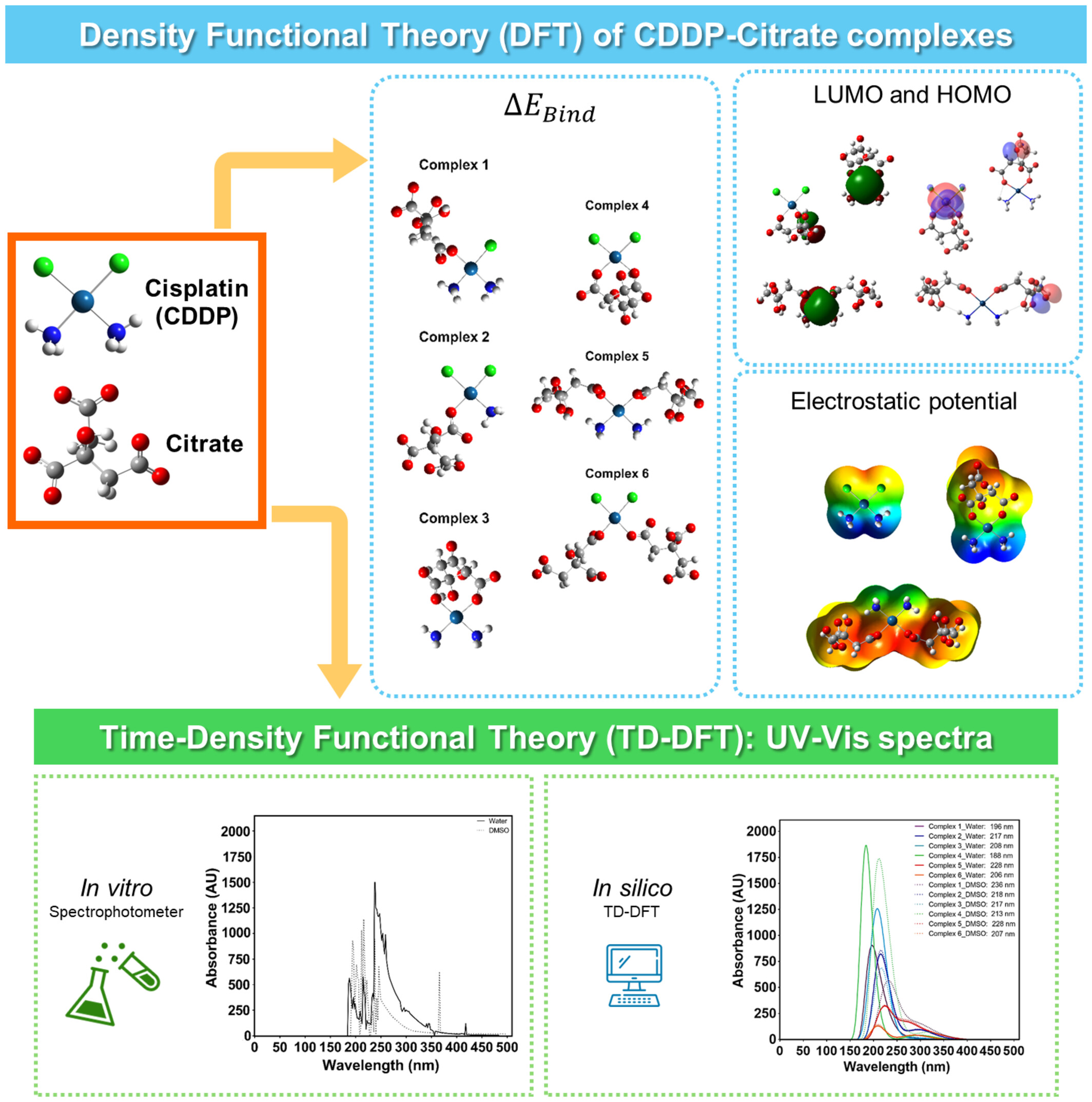
Nanoparticles were developed in vitro and in silico to overcome limitations of cisplatin (CDDP), Platinum (Pt) based anticancer drug. Citrate is commonly biocompatible in nano-drug synthesis. In this study, density functional theory (DFT) has been investigated to study the physicochemical and electronic properties of six CDDP-citrate complexes in water and DMSO. The frontier orbitals, electron densities mapping, and electrostatic potential analysis sheds light on the effective complex for nanomedicine applications.
CGPDTA: An Explainable Transfer Learning-Based Predictor With Molecule Substructure Graph for Drug-Target Binding Affinity
- First Published: 09 December 2024
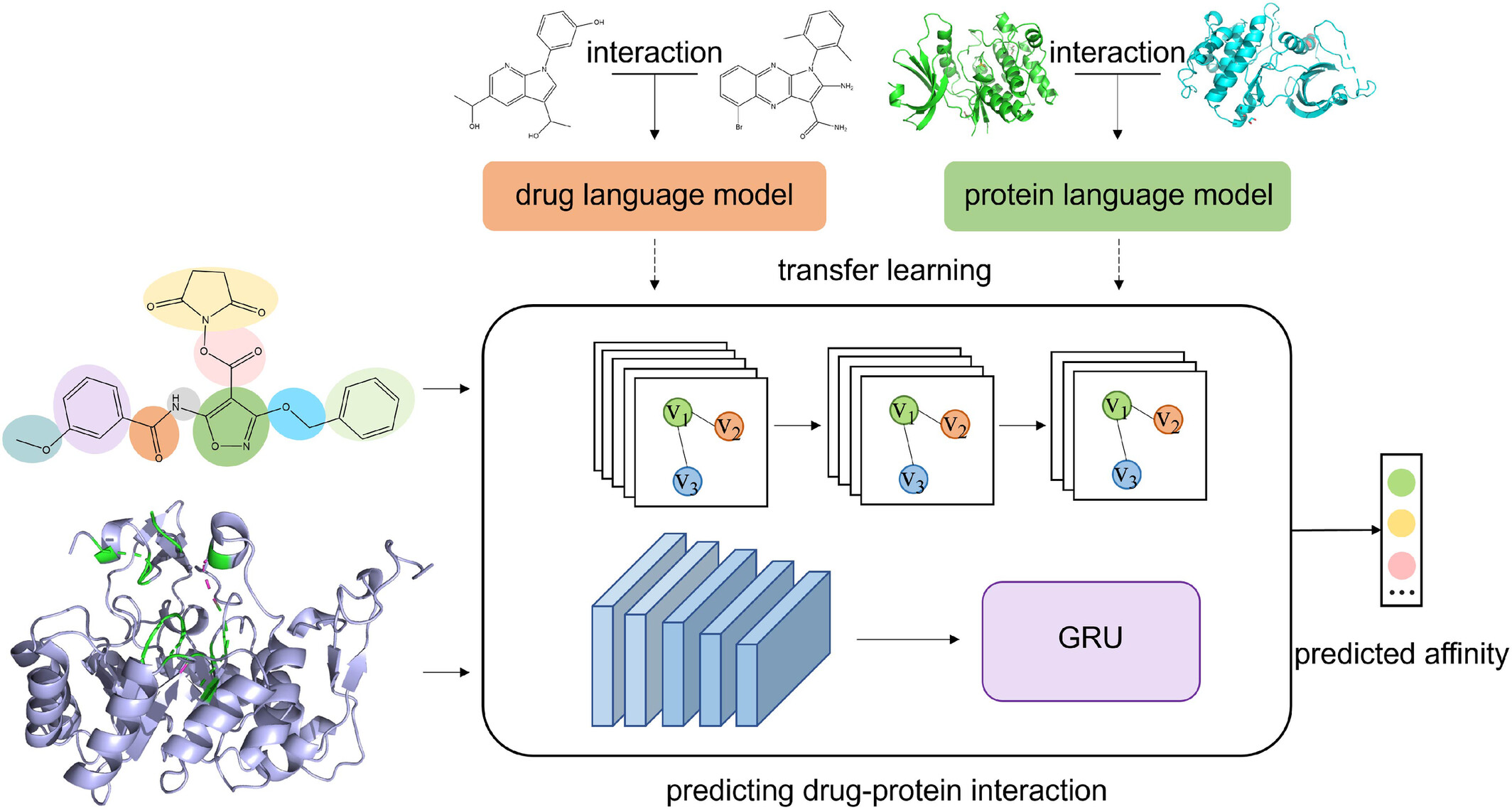
CGPDTA uses transfer learning to predict drug-target binding affinity by extracting effective drug-protein interaction information through the lens of a drug substructure graph.
Computer Folding of Parallel DNA G-Quadruplex: Hitchhiker's Guide to the Conformational Space
- First Published: 09 December 2024
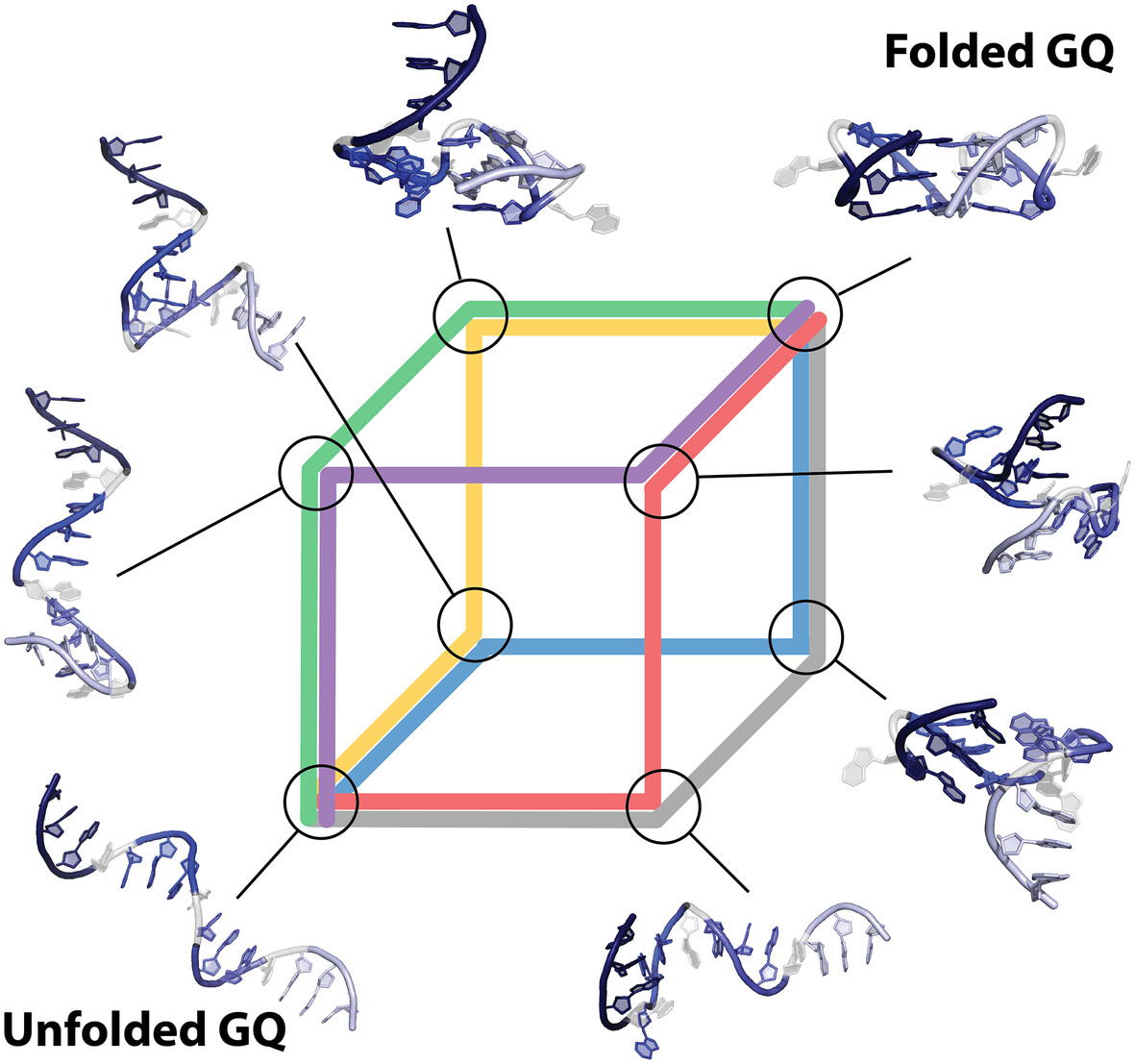
This study presents a novel computational protocol for folding guanine quadruplexes (GQs) from single-strand conformation to their native state. Utilizing two enhanced sampling techniques, the protocol models multichannel folding pathways, elucidating diverse intermediates and mechanisms. Despite its focus on the all-anti parallel GQ topology, this approach significantly enhances our understanding of GQ stability and dynamics and can be adapted for more complex folding systems.
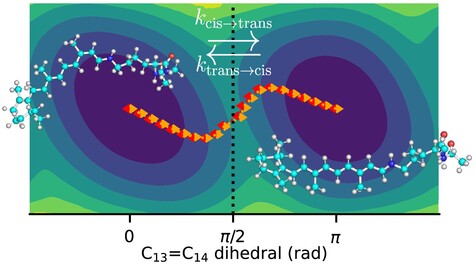
Accuracy of Reaction Coordinate Based Rate Theories for Modelling Chemical Reactions: Insights From the Thermal Isomerization in Retinal
- First Published: 10 December 2024
Improving the Efficiency of Electrostatic Embedding Using the Fast Multipole Method
- First Published: 10 December 2024
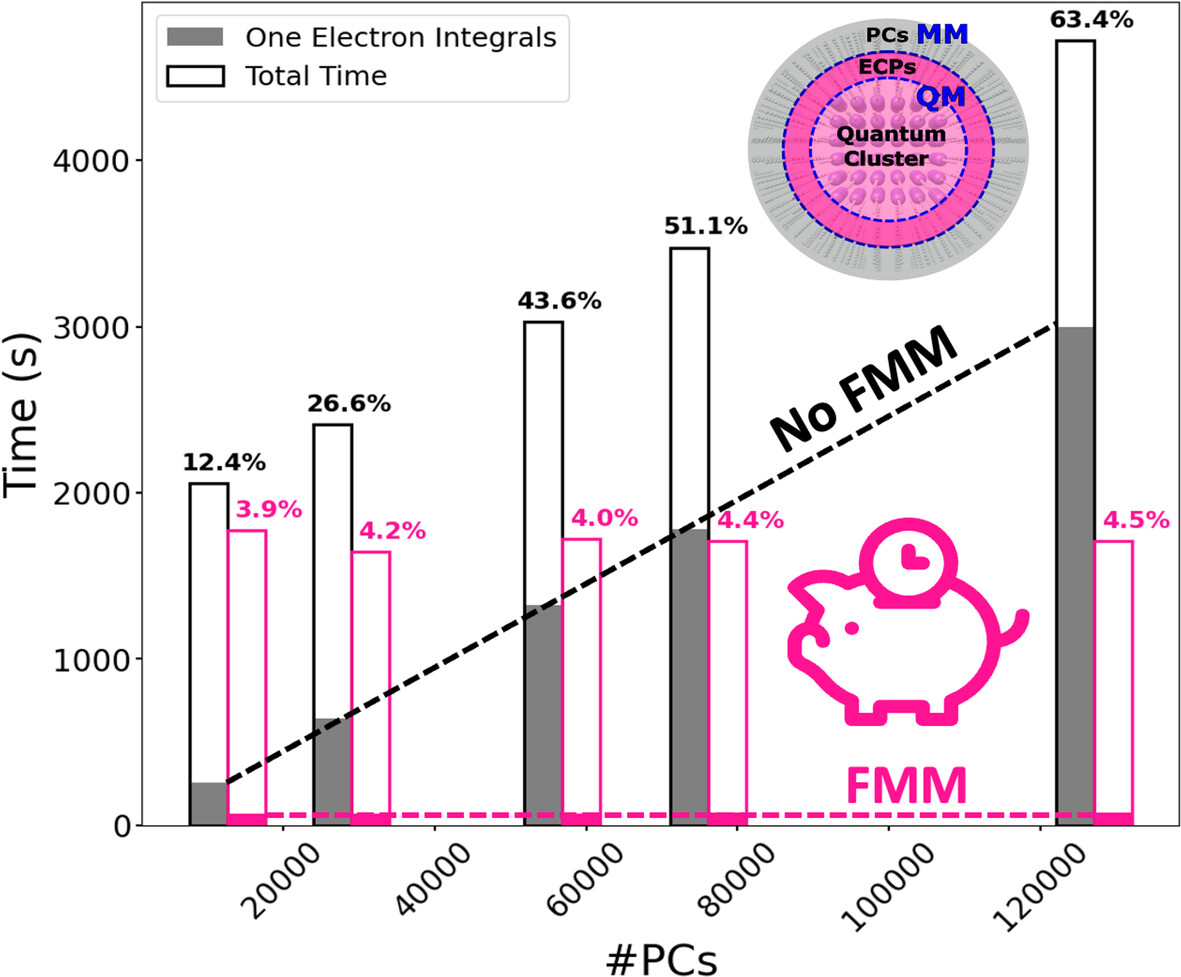
This paper reports the improvement in the efficiency of Embedded Cluster Model (ECM) calculations in ORCA thanks to the implementation of the Fast Multipole Method. The outcome of the work is a one order of magnitude minimum speedup for the evaluation of the electrostatic potentials.
pKa prediction in non-aqueous solvents
- First Published: 11 December 2024
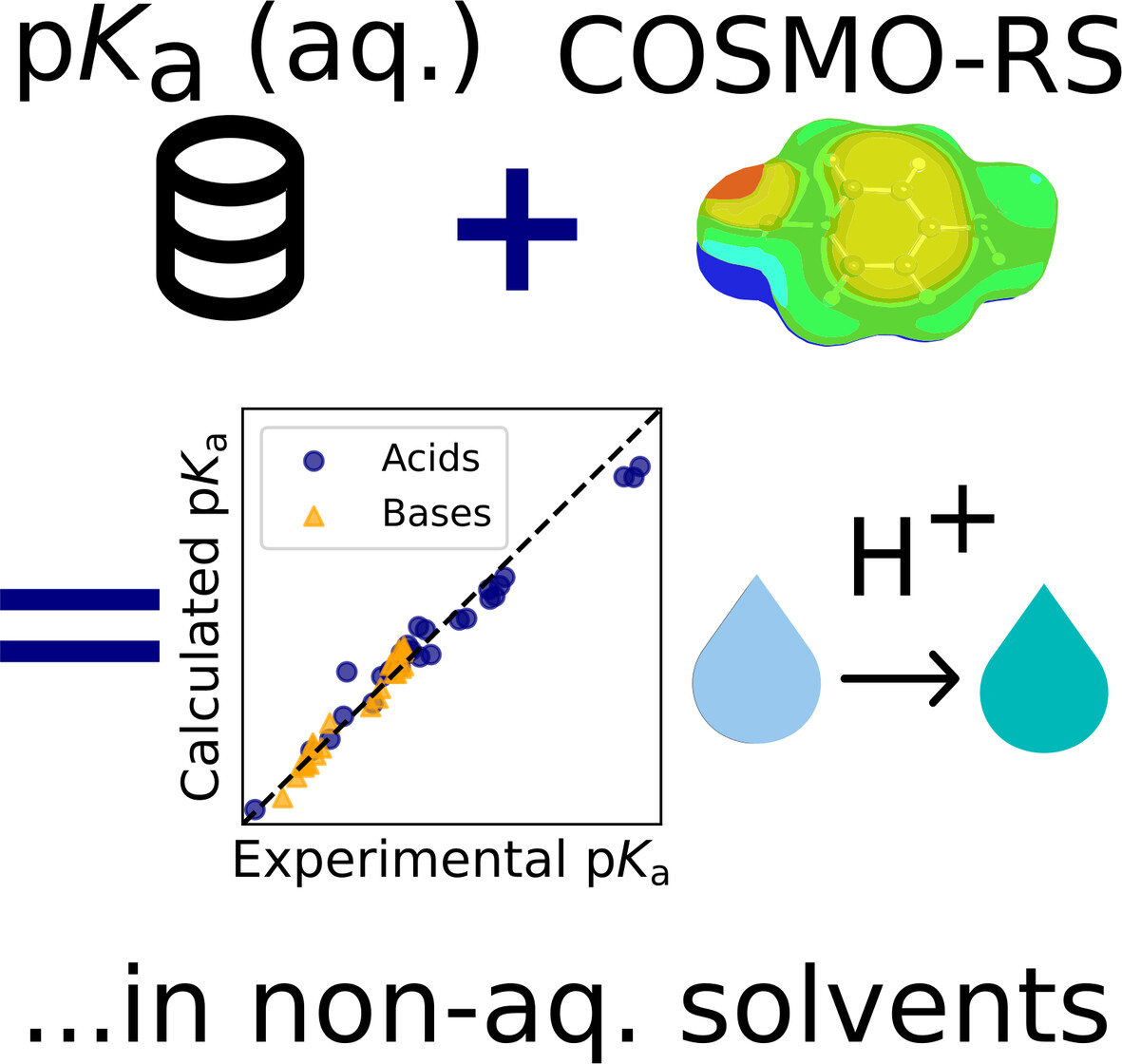
The acid dissociation constant (pKa) plays a crucial role in applications ranging from pharmaceutical synthesis to environmental science. This work demonstrates that popular solvation model COSMO-RS can be used in conjunction with aqueous pKa data to predict the pKa in many non-aqueous solvents, which can then be leveraged to approximate the transfer free energy of the proton between solvents. This work also explores inconsistencies in experimental data that sometimes lead to worse model agreement.
The Influence of the Solvation on the Bonding of Molecular Complexes of Diatomic Halogens With Nitrogen-Containing Donors and Their Stability With Respect to the Heterolytic Halogen-Halogen Bond Splitting
- First Published: 13 December 2024
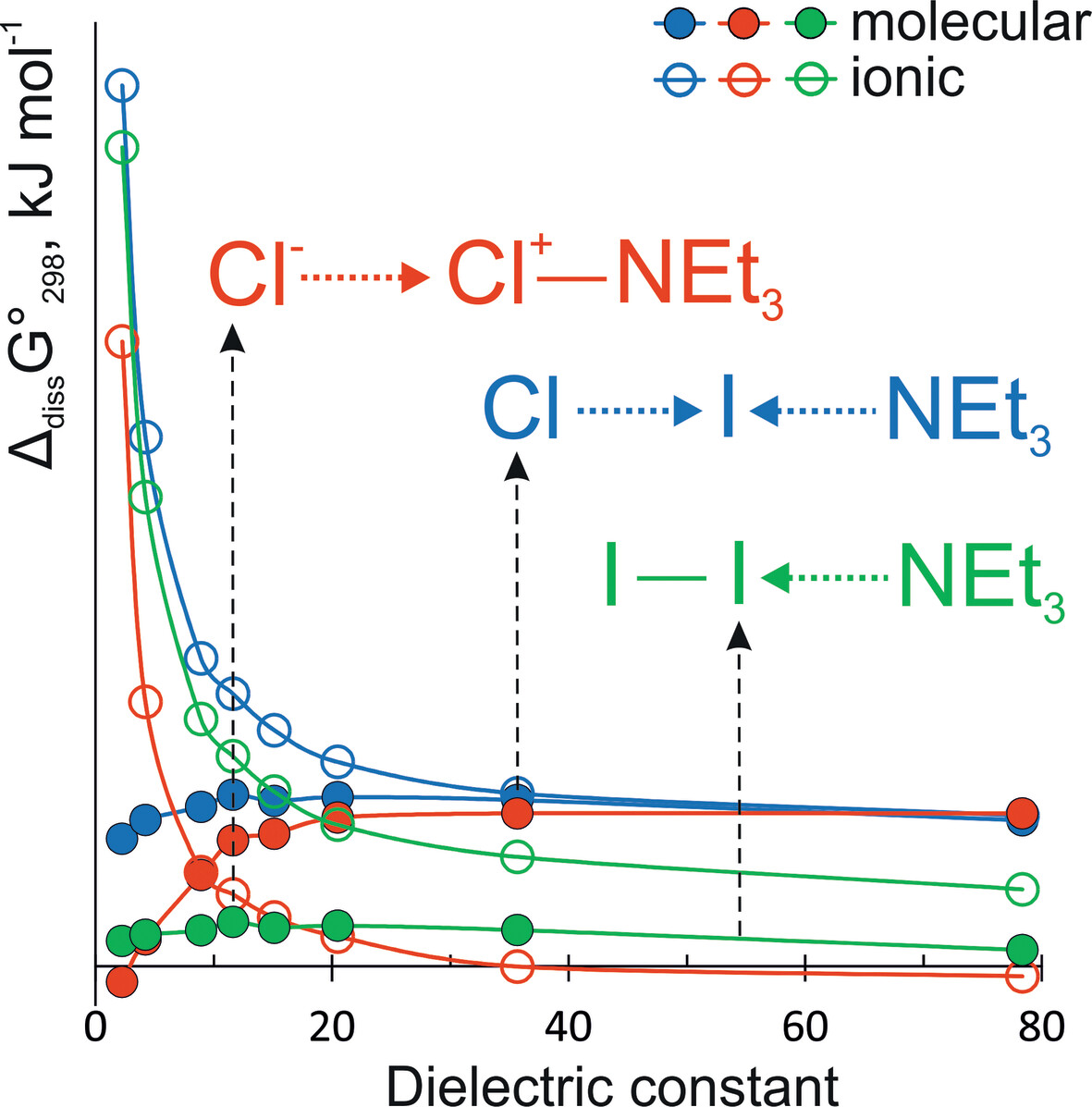
Increase of the solvent polarity leads to changes in the chemical bonding from molecular complexes to 3-center 4-electron bonds and to the contact ion pairs, facilitating heterolytic halogen-halogen bond splitting. Structural changes upon solvation in polar solvents are significant and single point solvation energy computations on gas phase optimized geometries are not reliable for a such systems.
Ultrafast Dynamics of Diketopyrrolopyrrole Dimers
- First Published: 14 December 2024

Using nonadiabatic dynamics, we uncover an ultrafast hydrogen migration-driven internal conversion pathway in diketopyrrolopyrrole dimers with a sub-picosecond excited-state lifetime. This work highlights the critical impact of state intersections on photophysical performance, offering insights for functionalizing DPP systems to suppress nonradiative decay.
CoTCNQ as a Catalyst for CO2 Electroreduction: A First Principles r2SCAN Meta-GGA Investigation
- First Published: 16 December 2024
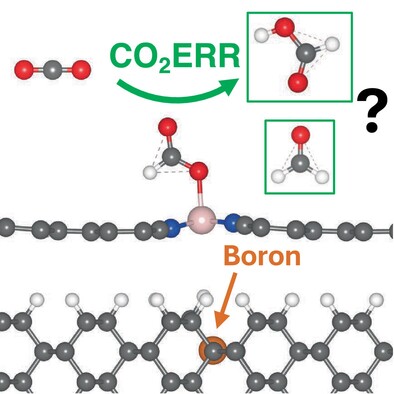
We investigate, using DFT, cobalt-coordinated tetracyanoquinodimethane (R-CoTCNQ) as a metal–organic framework catalysis for the CO2 electroreduction reaction. We find that reduced (charged) R-CoTCNQ may produce formic acid and/or formaldehyde reaction products as both a supported and unsupported catalyst.
SOFTWARE NOTE
Groupy: An Open-Source Toolkit for Molecular Simulation and Property Calculation
- First Published: 16 December 2024
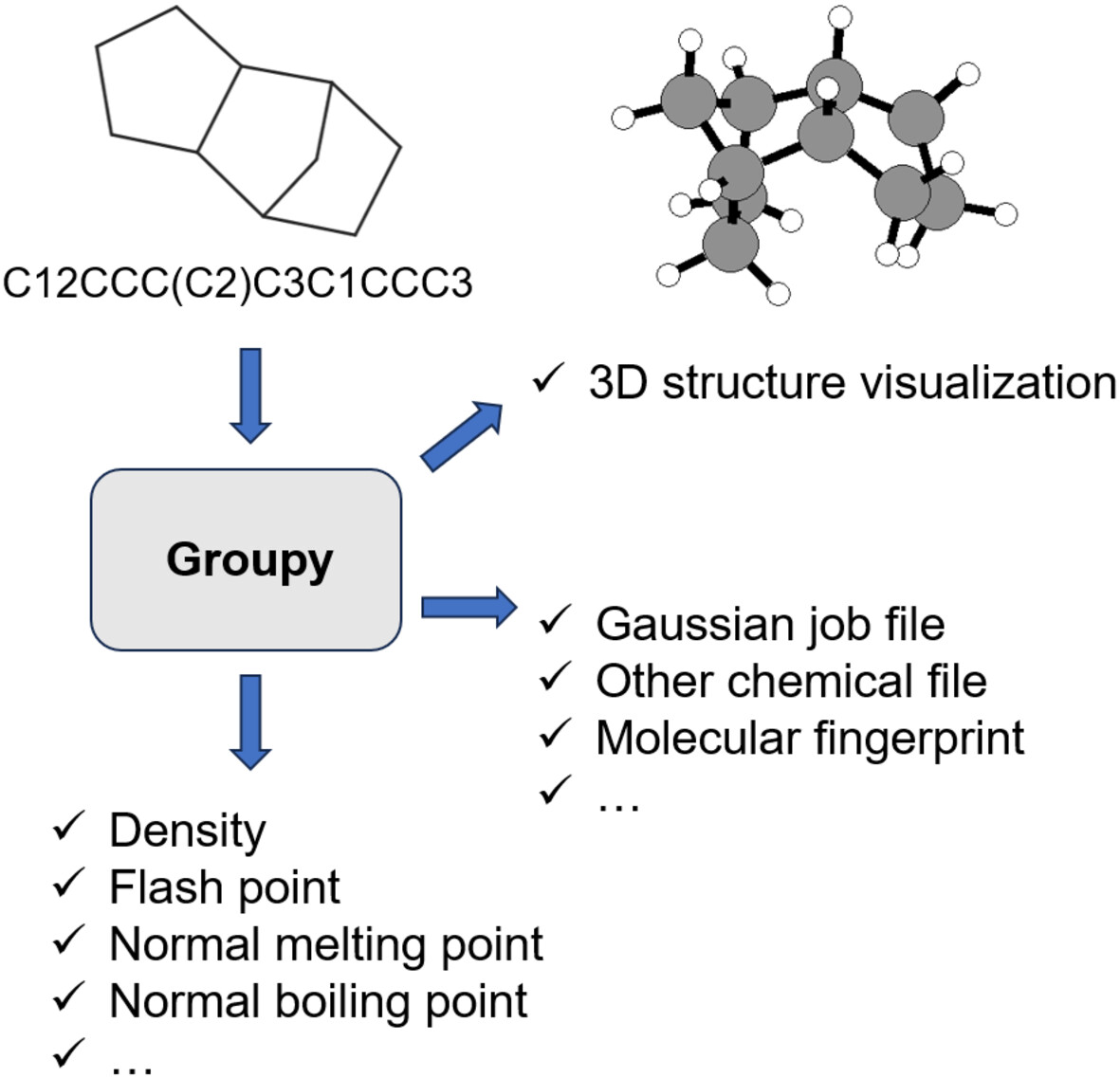
Groupy is a package targeted for calculating different molecular properties such as density, normal boiling point, normal melting point, critical temperature, flash point, critical pressure, critical volume, Gibbs free energy, enthalpy of formation, enthalpy of fusion, and enthalpy of vaporization. Groupy also can serve as an intermediary platform that can create input files for Gaussian and visualize 3D structure of a molecule based on SMILES.
RESEARCH ARTICLE
Tuning Electronic Relaxation of Nanorings Through Their Interlocking
- First Published: 16 December 2024
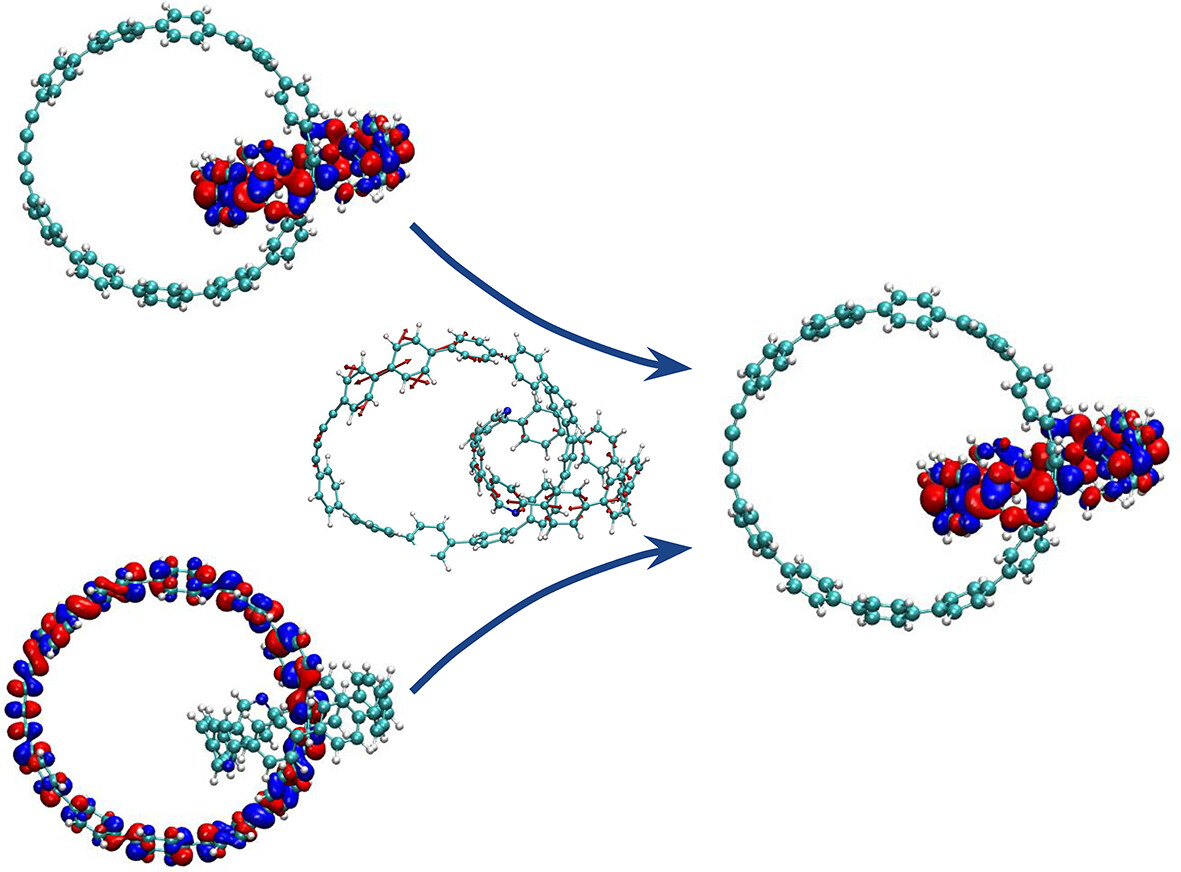
All-benzene catenanes experience faster relaxations than individual units. They present overlapping energy manifolds that include electronic excited states spatially localized on the different moieties, increasing the density of states. This result suggests a viable strategy for tuning the internal conversion rates in a quest for their utilization for new applications.
MARVEL Analysis of High-Resolution Rovibrational Spectra of 16O13C18O
- First Published: 19 December 2024
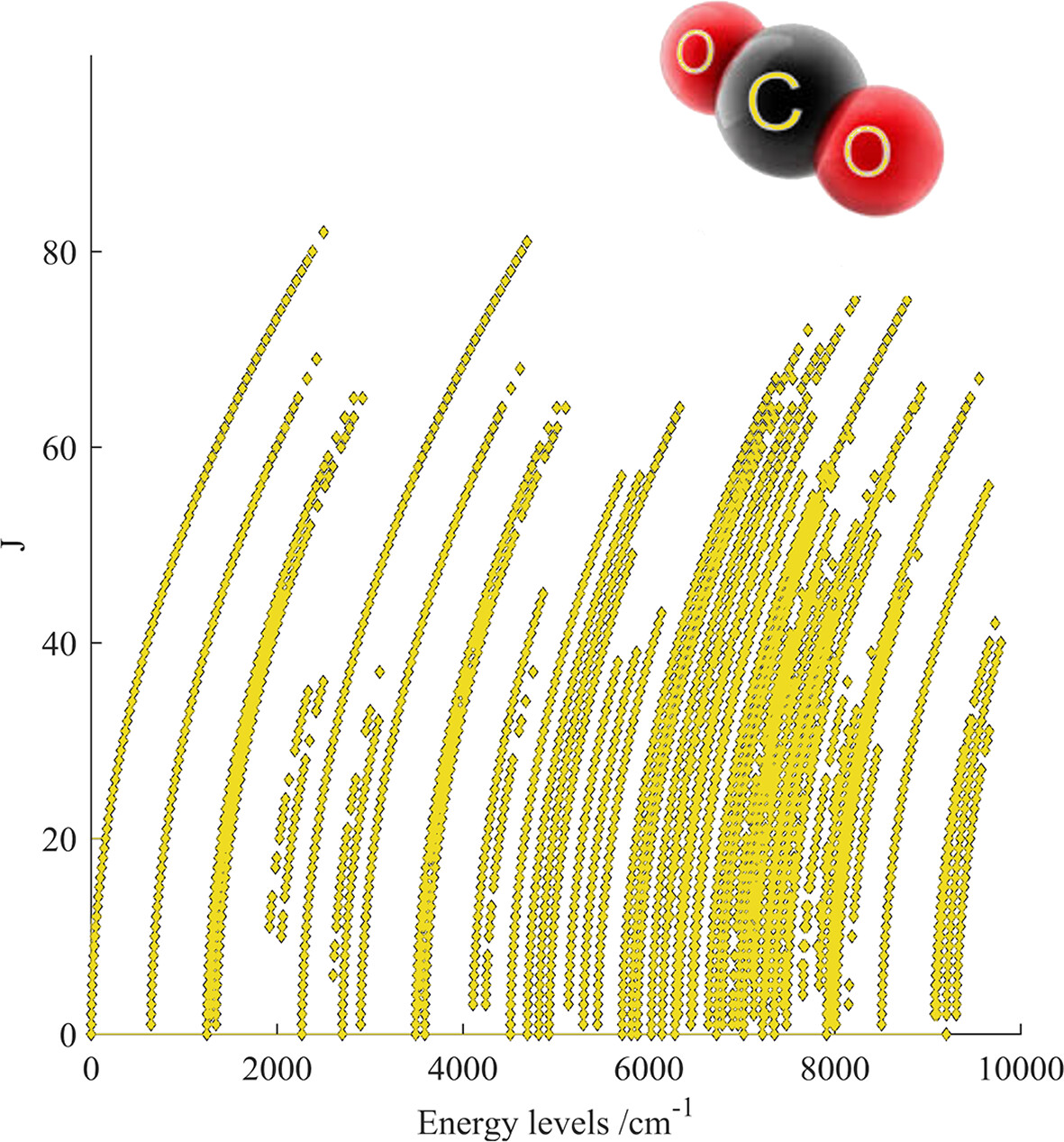
12348 measured transitions in the wavenumber range 578–9318 cm−1 for carbon dioxide (isotopologue 638) were used° to calculating 3975 empirical rovibrational energy levels with J° up to 82.
Additive CHARMM Force Field for Pterins and Folates
- First Published: 22 December 2024
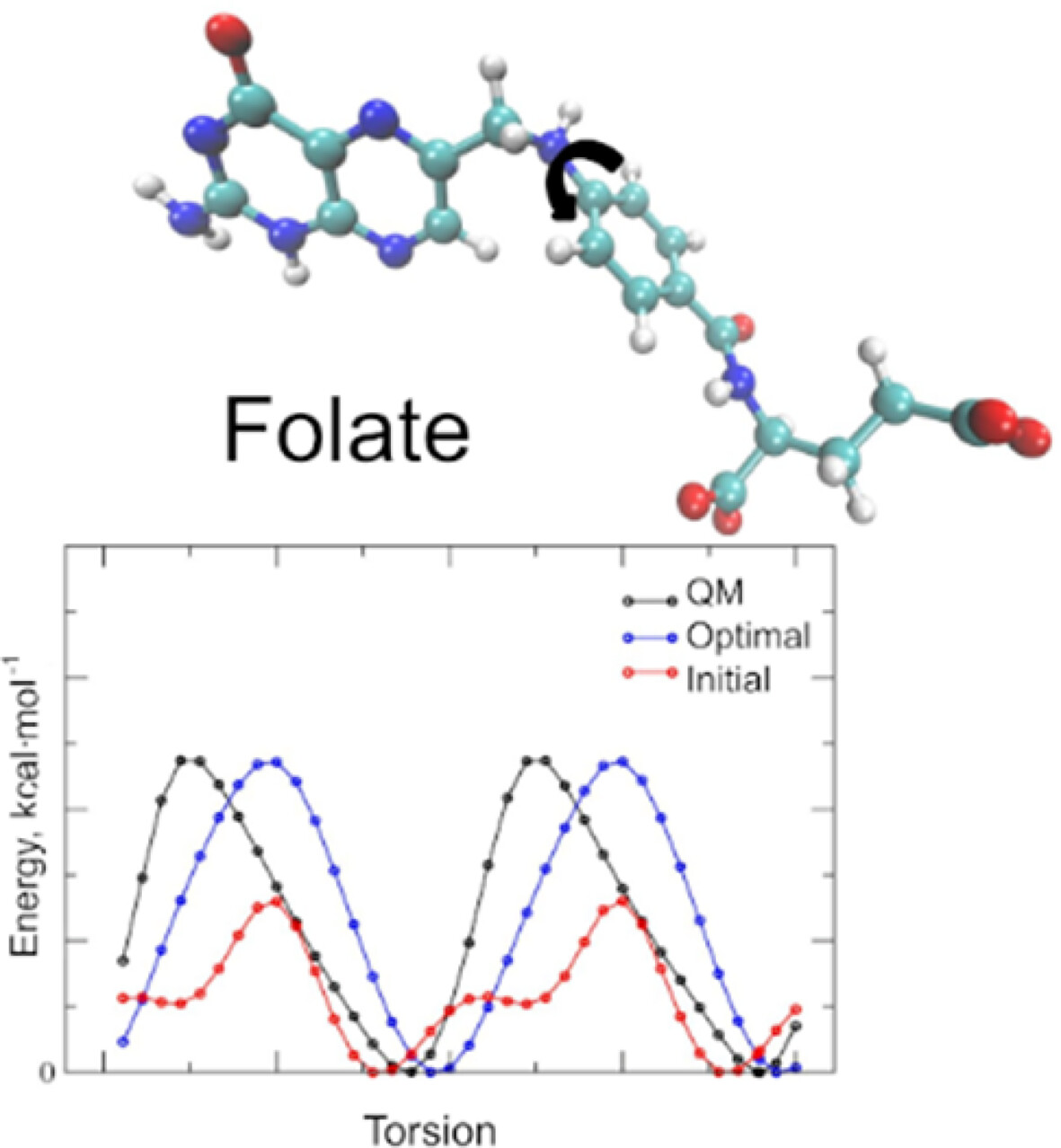
Folates, essential cofactors in one-carbon metabolism, play a pivotal role in critical physiological processes, including biosynthesis, epigenetic regulation, and redox defense. This study presents a molecular mechanics model for folates, parameterized for compatibility with the CHARMM36 all-atom force field. The model includes 18 folate derivatives across multiple redox and protonation states, employing quantum mechanical reference data to derive partial charges and optimize bonded terms. Simulations of protein complexes validated the improved accuracy of the new parameters in capturing folate–protein interactions. This model allows studying folate dynamics and serves as a foundation for further parameterization of folate derivatives.
Computing Accurate & Reliable Rovibrational Spectral Data for Aluminum-Bearing Molecules
- First Published: 23 December 2024
The F12-TcCR+TZ QFF alleviates many of the issues present in other QFF + VPT2 anharmonic frequency computations for aluminum-bearing molecules. This allows for more accurate computational spectroscopic data for atmospheric and interstellar detection of these aluminum-bearing molecules.
A Polarizable CASSCF/MM Approach Using the Interface Between OpenMMPol Library and Cfour
- First Published: 24 December 2024
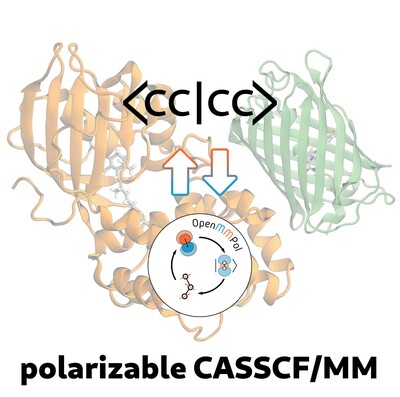
Including mutual polarization effects in quantum mechanics/molecular mechanics (QM/MM) approaches is important especially when excited states are involved. Here we present a polarizable embedding QM/MM model that couples Complete Active Space Self-Consistent Field (CASSCF) descriptions and the AMOEBA polarizable force field. The method is tested on two photoreceptor proteins for which we simulate the excitation process through geometry optimization of the ground state and calculation of the vertical excitation energy
VDAC Solvation Free Energy Calculation by a Nonuniform Size Modified Poisson–Boltzmann Ion Channel Model
- First Published: 26 December 2024
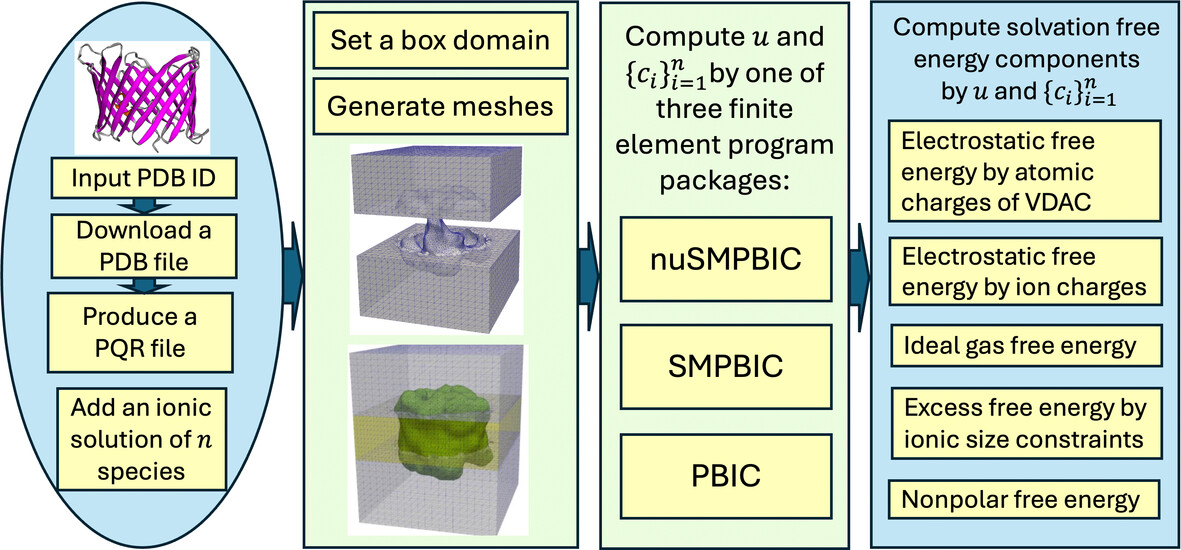
Numerical schemes are developed to compute the five solvation free energy components in terms of one electrostatic potential, and ionic concentration functions , calculated by our nuSMPBIC, SMPBIC, and PBIC finite element packages. The schemes are then integrated with our improved mesh generation package, the PDB2PQR package for generating PQR files, and the OPM database for downloading PDB files, resulting in the voltage-dependent anion channel solvation free energy calculation package.
Advanced Computational Insights Into Cs₂NaScX₆ (X = Cl, Br) ₆ Double Perovskites: Structural Stability, Elastic Properties, and Optical Characteristics for Next-Generation Photovoltaics
- First Published: 24 December 2024

We investigate the structural, electronic, optical, and elastic properties of Cs₂NaScX₆ (X = Cl, Br) double perovskites using DFT in WIEN2k. The compounds exhibit cubic symmetry, mechanical stability, ductile behavior, and pronounced visible-light absorption, with band gaps of 3.138 eV and 3.977 eV, respectively. These findings highlight their potential for advanced photovoltaic applications.
Comprehensive Analysis of Deuterium Isotope Effects on Ionic H3O+…π Interactions Using Multi-Component Quantum Mechanics Methods
- First Published: 20 December 2024
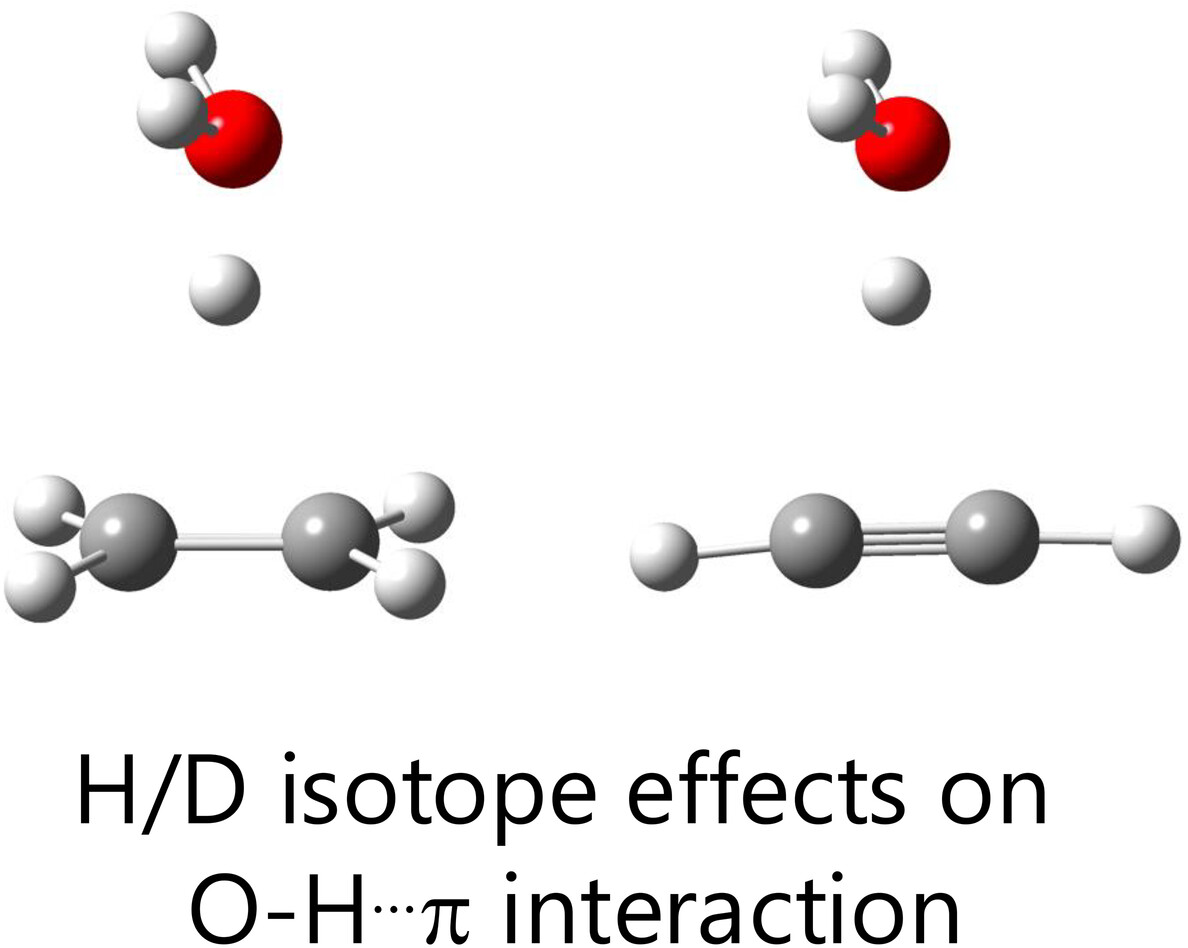
Deuterium isotope effects on interaction energies and geometrical parameters in ionic H3O+…π interactions were comprehensively analyzed using multi-component quantum mechanics. Replacing H3O+ with D3O+ reduced interaction energies and induced changes in hydrogen-bonded distances. Natural energy decomposition analysis revealed strong correlations between H/D isotope effects on these distances and energy components.
Does Basis Set Superposition Error Significantly Affect Post-CCSD(T) Corrections?
- First Published: 24 December 2024
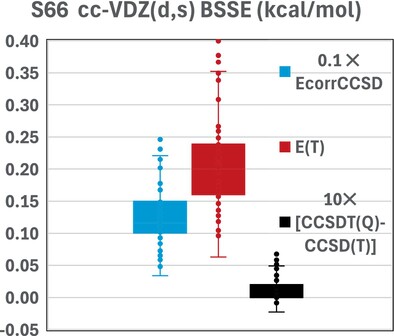
As can be seen in the above box plot, the counterpoise correction on the post-CCSD(T) corrections is over an order of magnitude smaller than that on the connected triples, and two orders smaller than that on the CCSD correlation energy.
Influence of Ligand Complexity on the Spectroscopic Properties of Type 1 Copper Sites: A Theoretical Study
- First Published: 26 December 2024
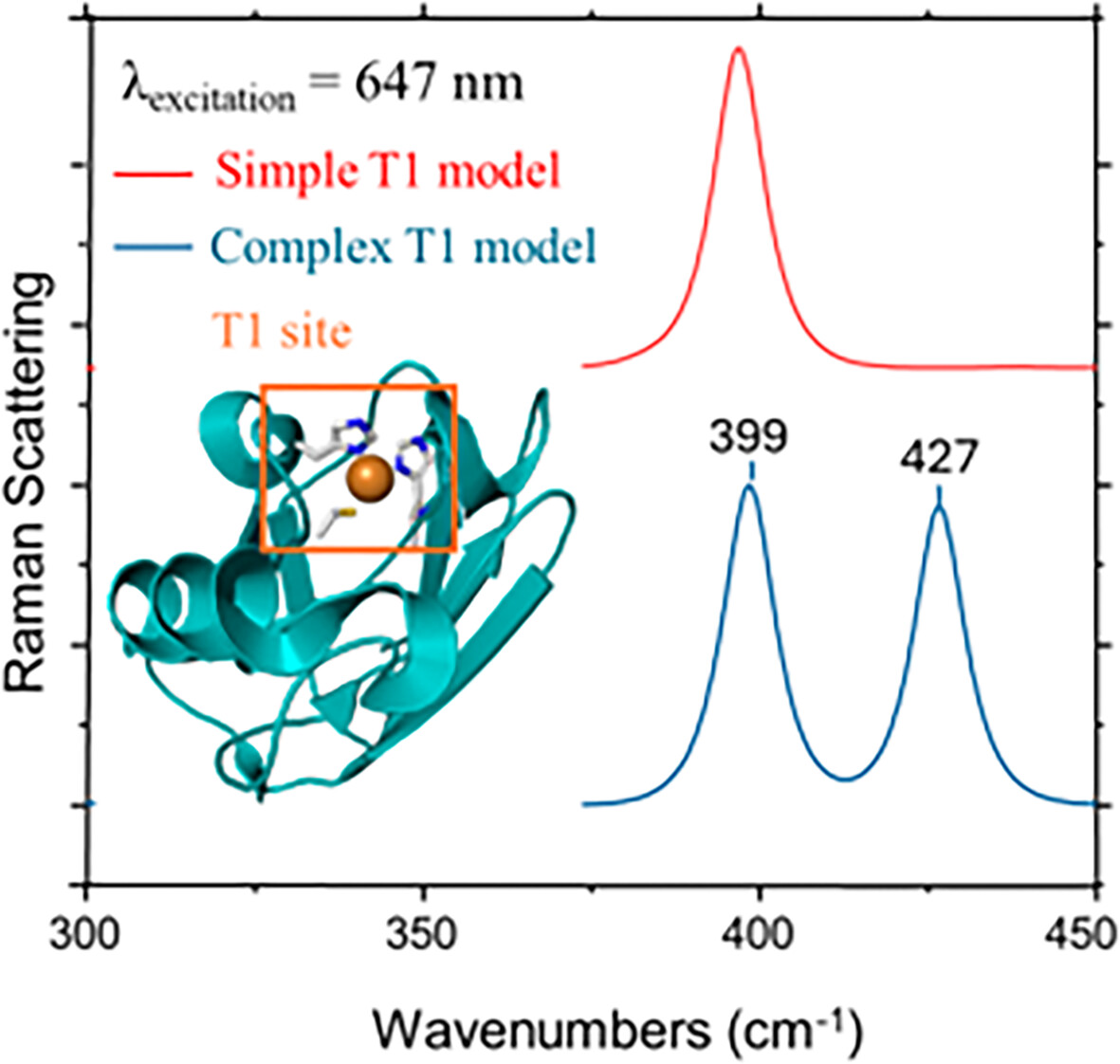
Calculated resonance Raman spectra (647 nm excitation) for a Type 1 (T1) copper site, comparing simplest and most complex models. The simplest model exhibits one strong band, while the complex model shows band splitting, demonstrating how increased model sophistication affects predicted vibrational spectra of copper proteins.
Machine Learning Prediction of Physicochemical Properties in Lithium-Ion Battery Electrolytes With Active Learning Applied to Graph Neural Networks
- First Published: 26 December 2024
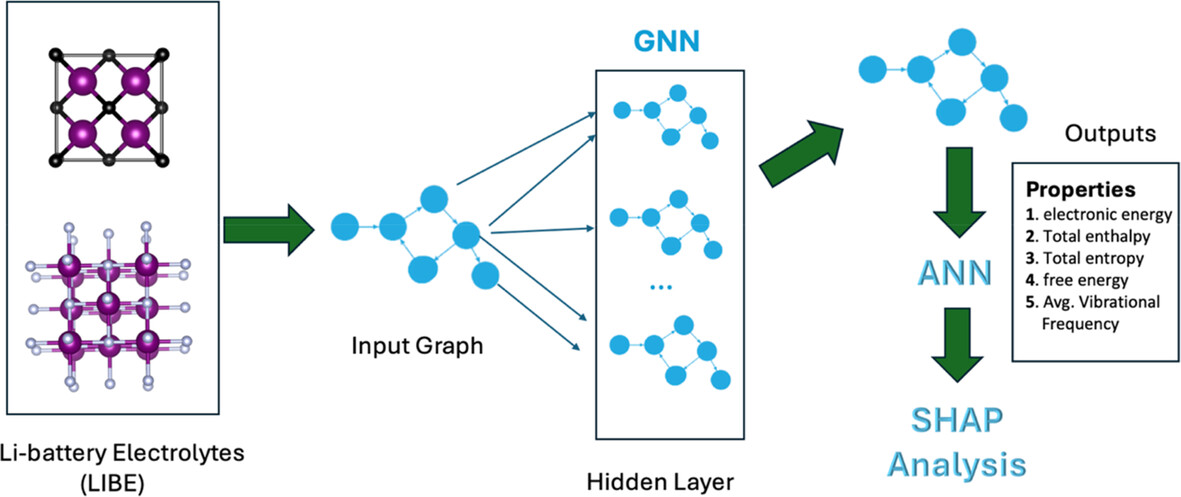
Advanced machine learning methods could be employed to model key properties, including electronic energy and vibration frequencies, of the Lithium-ion battery electrolytes.
Machine Learning-Corrected Simplified Time-Dependent DFT for Systems With Inverted Single-t-o-Triplet Gaps of Interest for Photocatalytic Water Splitting
- First Published: 30 December 2024

We present a delta machine learning (Δ-ML) tool that corrects simplified Tamm–Dancoff Approximation excited state energies to produce second order algebraic diagrammatic construction (ADC(2))-like results. We use Δ-ML to screen aza-triangulenes to find those with inverted singlet-to-triplet-gaps (INVEST) and have excited states suitable for photocatalytic water splitting (PWS). Our Δ-ML model has an 85% recall for systems identified by ADC(2) as having both the INVEST and PWS properties, producing MADs within 28–50 meV of ADC(2).
A Closer Look at the FeS Heme Bonds in Azotobacter vinelandii Bacterioferritin: QM/MM and Local Mode Analysis
- First Published: 03 January 2025
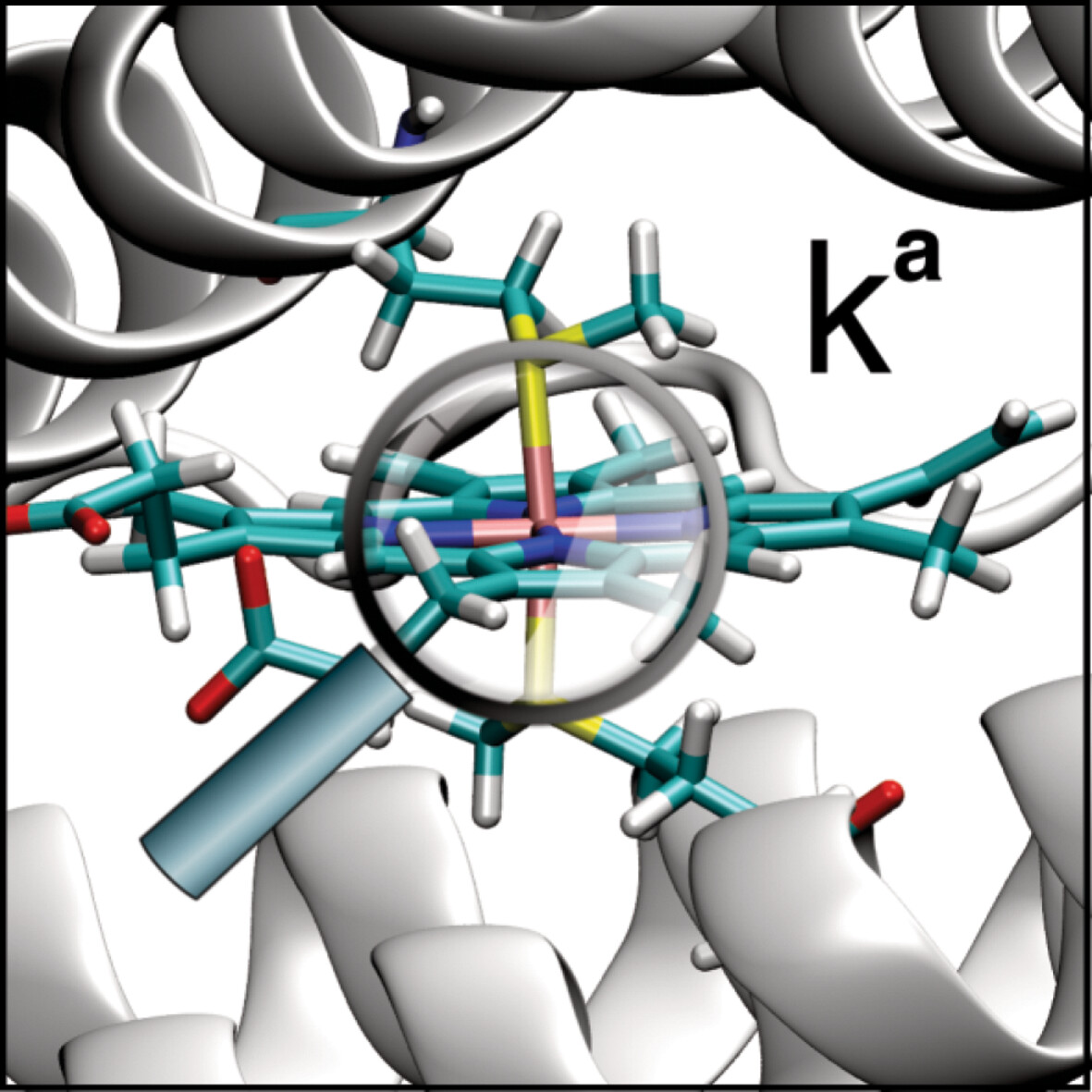
Using the QM/MM method, we analyzed the strength of FeS bonds in heme groups of oxidized and reduced forms of bacterioferritin. The strength of FeS bonds was correlated with bond length, energy density at bond critical point, and charge difference between F and S atoms. The change of oxidation state from ferrous to ferric, makes FeS bonds weaker, longer, more covalent, and more polar. We have also found that stronger FeS bonds correlate with stronger SFeS bond bending.
Examining the Impact of Local Constraint Violations on Energy Computations in DFT
- First Published: 02 January 2025
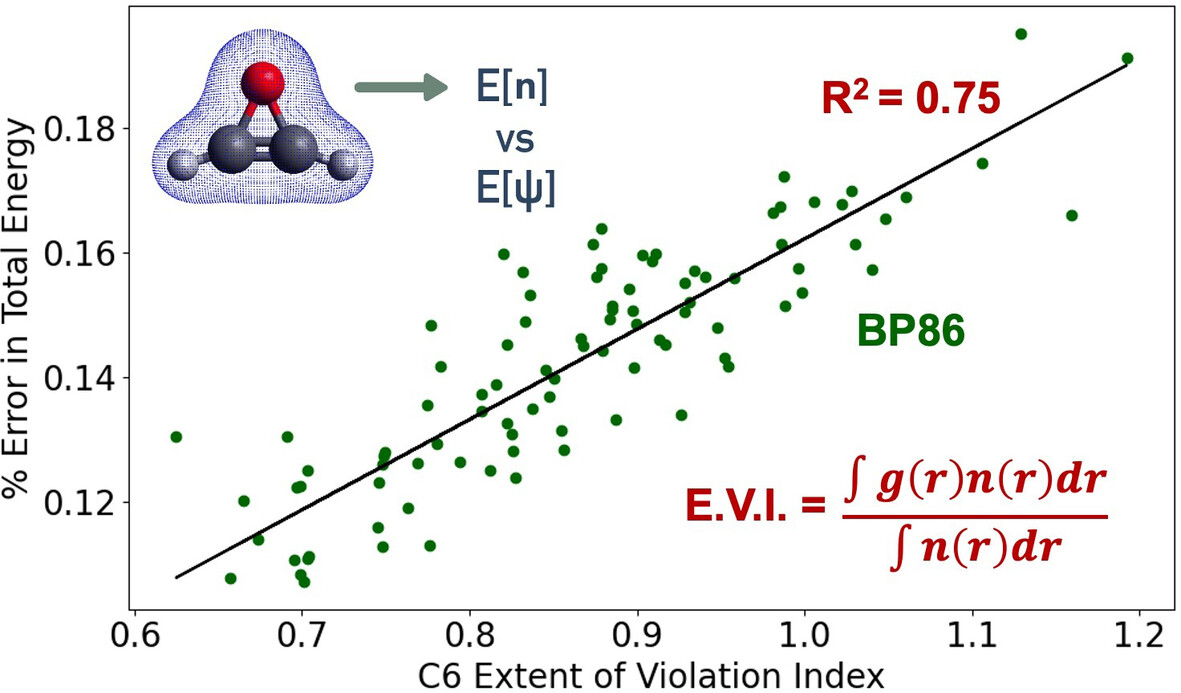
Local constraint violations by the BP86 functional, quantified by our metric, trending with total energy errors: molecules with more violations also have higher errors in DFT total energies.
Thermodynamic Stability in Transition Metal-Hydrogen Dications: Potential Energy Curves, Spectroscopic Parameters, and Bonding for VH2+
- First Published: 04 January 2025
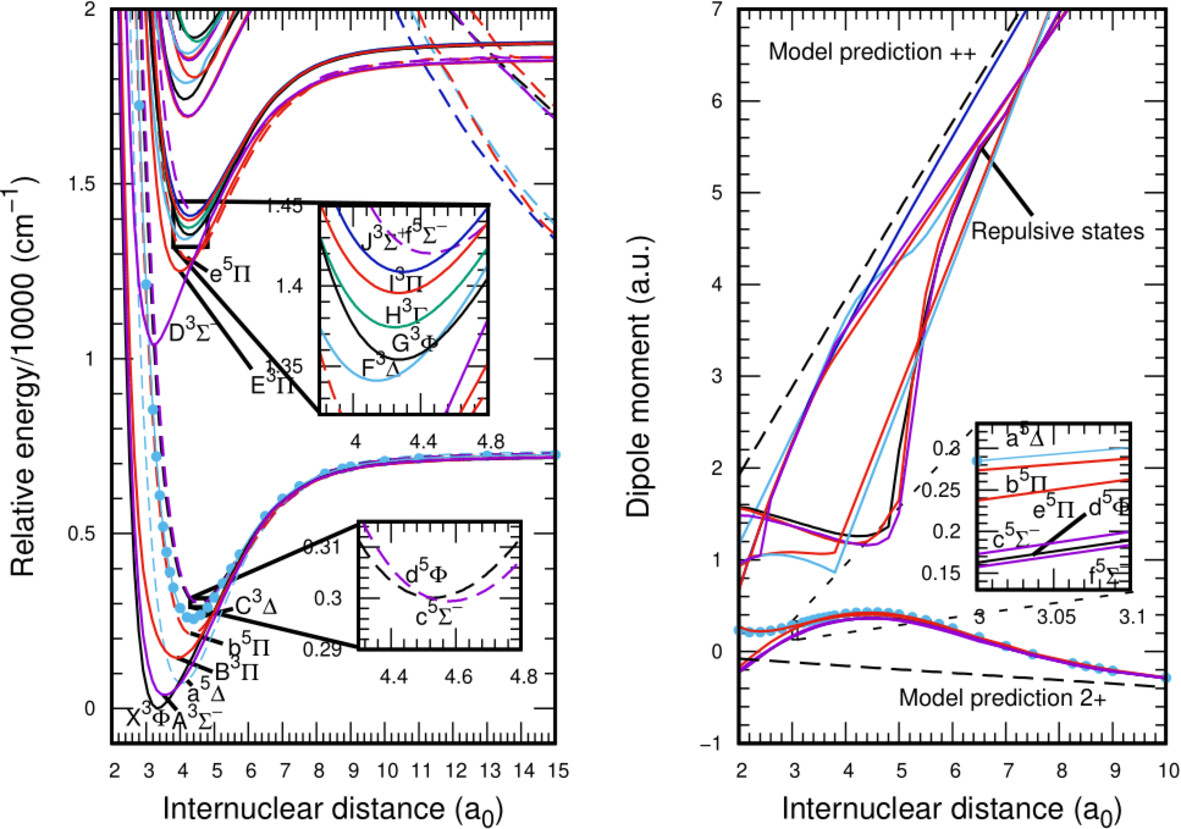
Thermodynamic stability in transition metal-containing diatomic dication—VH2+.
Appraisal of the Fragments-In-Fragments Method for the Energetics of Individual Hydrogen Bonds in Molecular Crystals
- First Published: 04 January 2025
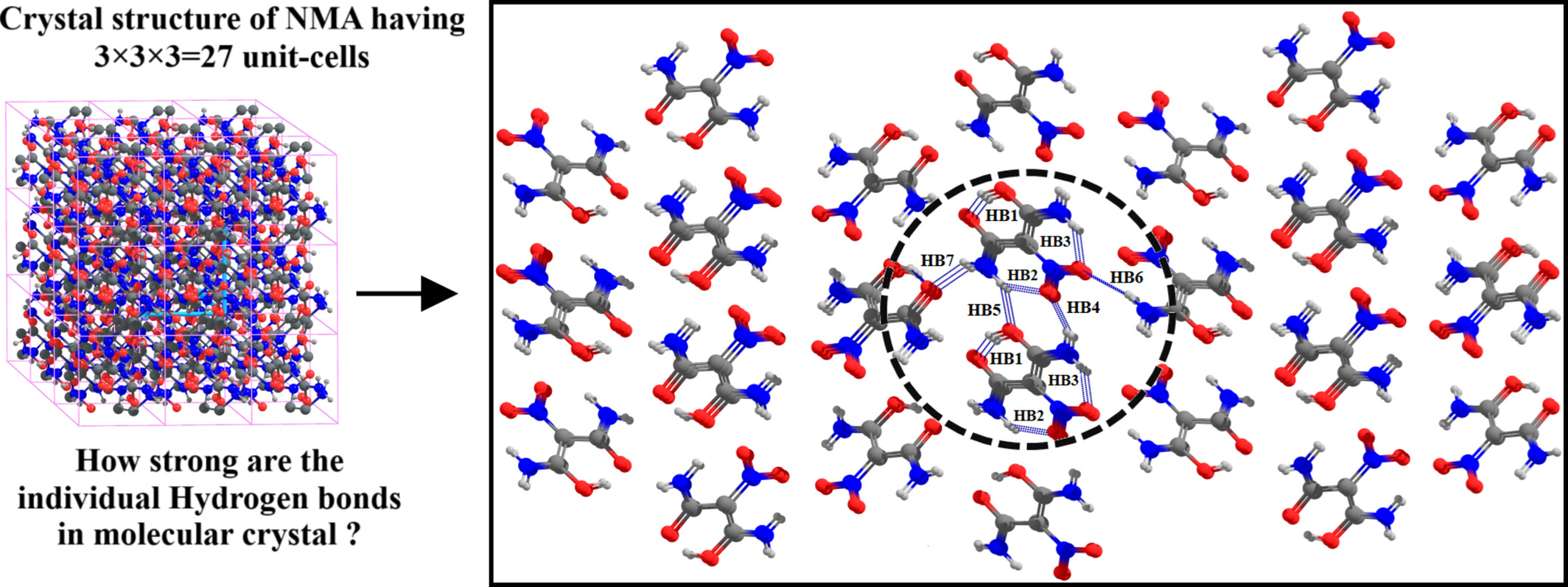
The individual hydrogen bond energies in molecular crystal of nitromalonamide and salicylic acid are calculated by the MTA-based and the Frags-in-Frags methods. The direct application of MTA-based method is very expensive and may not be possible with off-the-shelf-hardware. The Frags-in-Frags method provides accurate results and is inexpensive.
Practical Machine Learning Strategies. I. Correcting the MMFF Molecular Mechanics Model to More Accurately Provide Conformational Energy Differences in Flexible Organic Molecules
- First Published: 05 January 2025
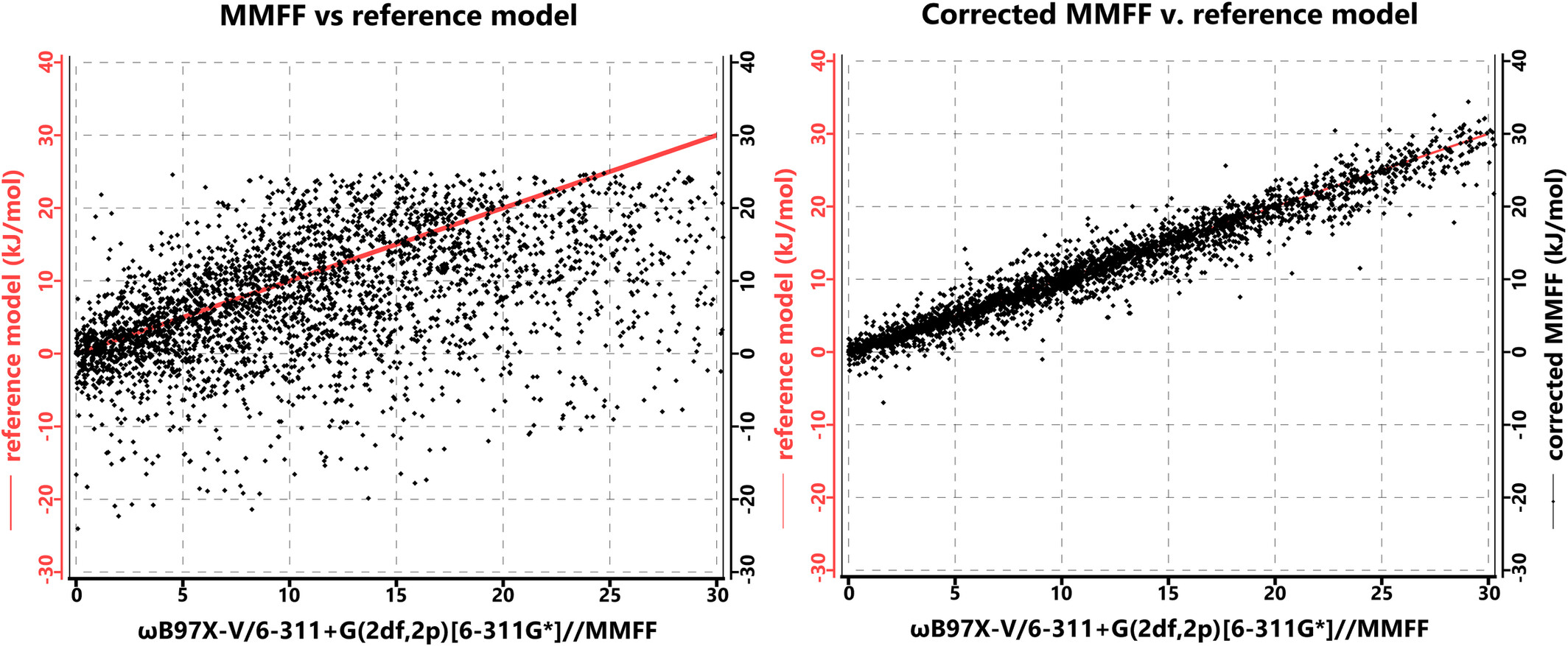
A neural net has been trained to fit conformer energy differences obtained from large basis set density functional calculations. Applied as a correction to the MMFF molecular model, this approach leads to dramatic improvement with a small (insignificant) increase in calculation cost.
Sign up for email alerts
Tools
