Journal list menu

 Issue
IssueAngewandte Chemie: Volume 103, Issue 12
cpi-A421, 1559-1743Dezember 1991
Export Citations
Download PDFs
Titelbild
Impressum
Graphisches Inhaltsverzeichnis
Aufsätze
Neue Wege zur Nutzung von Aminosäuren als chirale Bausteine in der organischen Synthese†
- Pages: 1559-1573
- First Published: Dezember 1991
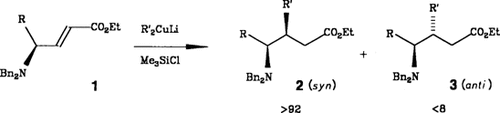
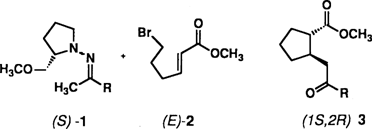
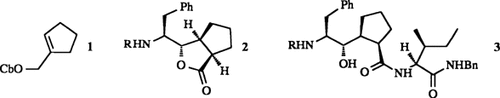
Schier unerschöpflich scheint das Synthesepotential von Aminosäuren. Am Stickstoff unterschiedlich geschützte α-Aminosäuren lassen sich in die entsprechenden α-Aminoaldehyde racemisierungsfrei überführen. Wählt man die richtigen Schutzgruppen („Schutzgruppen-Tuning”︁), z.B. zwei Benzylreste am Stickstoffatom, so sind Grignard-artige Reaktionen, Aldol- und Me3SiCN-Additionen sowie Hetero-Diels-Alder-Reaktionen erstmals unter hoher Nicht-Chelat-Kontrolle möglich. Durch Wittig-Reaktion können die α-Aminoaldehyde in elektronenarme γ-Aminoolefine umgewandelt werden, die ihrerseits stereoselektive Cuprat-, Michael- und Cycloadditionen eingehen. Die Umsetzung 1 → 2 + 3 ist ein Beispiel.
Volumeneffekt oder Drehtürmechanismus - schnelle Alkalimetall-Ionenleitung in Festkörpern mit rotationsfehlgeordneten komplexen Anionen†
- Pages: 1574-1586
- First Published: Dezember 1991
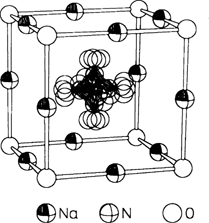
Transportphänomene im Festkörper sind für Grundlagenforschung und Anwendung gleichermaßen von Bedeutung. Besonderes Interesse wird dabei den „schnellen”︁ Ionenleitern wegen ihres Anwendungspotentials entgegengebracht. Auf der Basis von Ionenkristallen, in denen sich einerseits durch Dotierung hohe Ladungsträgerkonzentrationen - Punktdefekte in der Kationenteilstruktur - einstellen lassen, andererseits die Aktivierungsenergie des Platzwechsels durch translatorisch fixierte, jedoch rotatorisch bewegliche komplexe Anionen erniedrigt ist, wurden Alkalimetall-Ionenleiter mit hohen Leitfähigkeiten synthetisiert. Das Bild rechts zeigt einen Ausschnitt aus der Kristallstruktur der Hochtemperaturform von Na3NO3 ((NO2)ONa3).
1,4-Dihydropyridine: Einfluß von Chiralität und Konformation auf die Calcium-antagonistische und -agonistische Wirkung†
- Pages: 1587-1605
- First Published: Dezember 1991
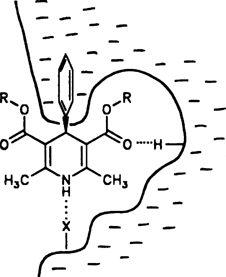
Die Therapie von Herz-Kreislauf-Erkrankungen ist ohne 1,4-Dihydropyridin-Derivate fast nicht mehr vorstellbar. Diese können als Calcium-Antagonisten oder Calcium-Agonisten wirken, d.h. den Ca2+-Einstrom in die Herz- und Gefäßmuskulatur hemmen bzw. steigern, wobei die Wirkungsweise entscheidend von der Konfiguration des Gesamtmoleküls und dem Substitutionsmuster am Pyridinring abhängt. Die Skizze rechts faßt die strukturellen Erfordernisse für eine effiziente Bindung eines typischen Calcium-Antagonisten an den Rezeptor zusammen. Ersetzt man in diesem Fall die linke Esterfunktion durch eine Nitrogruppe, so kehrt sich die pharmakologische Wirkung um.
Siliciumnitrid - vom Pulver zum keramischen Werkstoff†
- Pages: 1606-1625
- First Published: Dezember 1991
Nicht als Rohstoff zur technischen Herstellung von Ammoniak - wie um die Jahrhundertwende gedacht -, sondern als hochbelastbarer Werkstoff ist Siliciumnitrid heute von Interesse. So wird es im chemischen Apparatebau, in der Verschleiß- und Energietechnik, in der Metallbearbeitung und vor allem als Konstruktionsmaterial im Motoren- und Turbinenbau verwendet. Auf welchen chemischen Reaktionen die Si3N4-Herstellung basiert, welche Faktoren die Eigenschaften des Werkstoffs beeinflussen, und der Ablauf des technischen Verfahrens werden in dieser Übersicht vorgestellt.
Wege zu neuen aromatischen Polycarbonaten mit besonderen Werkstoffeigenschaften†
- Pages: 1626-1638
- First Published: Dezember 1991
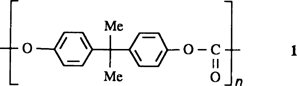
Wie das chemische und physikalische Potential der Polycarbonate optimal genutzt werden kann, um thermoplastische Hochleistungskunststoffe zu erhalten, schildert diese Übersicht. Die Hauptansatzpunkte sind, ausgehend vom Standardpolycarbonat 1, der Einbau anderer Monomere oder anderer Endgruppen, das Erzeugen von Verzweigungen, die Zugabe von Additiven und die Herstellung von Legierungen mit anderen Thermoplasten.
Glycolipide als Immunmodulatoren - Synthesen und Eigenschaften†
- Pages: 1639-1649
- First Published: Dezember 1991
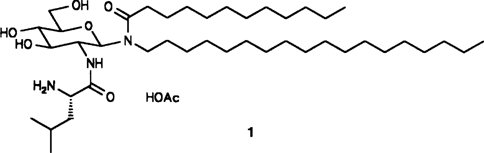
Eine Steigerung der spezifischen Immunantwort wird durch neuartige Glycolipidanaloga vom Typ des BAY R 1005 1 bewirkt. Diese sind lokal und systemisch gut verträglich; ihr Wirkungsmechanismus, der sich von dem bekannter Adjuvantien unterscheidet, läßt diese Substanzen für Immunisierungen von Patienten mit defekten T-Lymphozytenfunktionen, z.B. AIDS-Patienten, als besonders aussichtsreich erscheinen.
Herbizide in der Photosyntheseforschung†
- Pages: 1650-1663
- First Published: Dezember 1991
Eine Proteinuntereinheit im Photosystem II, das D1-Protein, ist der spezifische Angriffsort photosynthesehemmender Herbizide in Pflanzen. Wie genau die Vorstellungen über die Bindungsweise dieser Herbizide mittlerweile sind, ist im Bild rechts am Beispiel von Terbutryn, einem 1,3,5-Triazinderivat gezeigt. Es basiert auf Studien an herbizidtoleranten Mutanten, dem Einsatz von Techniken wie der Photoaffinitätsmarkierung sowie der engen strukturellen Verwandtschaft pflanzlicher und bakterieller Photosysteme. In diesem Fall lassen sich bereits Wechselwirkungen des Inhibitors mit spezifischen Aminosäuren der Bindungstasche diskutieren.
Zuschriften
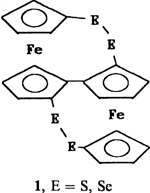
Derivate von 1,1″-Biferrocen mit Schwefel- und Selenbrücken†‡
- Pages: 1664-1665
- First Published: Dezember 1991
Über eine vierfach lithiierte Zwischenstufe ließen sich erstmals tetrasubstituierte Derivate von 1,1″-Biferrocen erhalten. Sie reagiert nicht nur beispielsweise mit Me3SiCl und MeSSMe, sondern auch mit Schwefel und Selen. In den Hauptprodukten dieser Reaktionen, 1, verklammern die Chalcogenide die beiden Ferroceneinheiten über E2-Brücken.
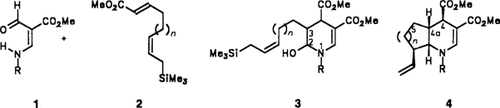
Stereoselektiver Aufbau anellierter Piperidine durch photochemische Cycloaddition und Iminium-Ion/Allylsilan-Cyclisierung†‡
- Pages: 1665-1667
- First Published: Dezember 1991
Nur eines von acht möglichen Diastereomeren entsteht bei der Umsetzung von Enamincarbaldehyden 1 (R H, CH3, p-MeOC6H5CH2) mit Alkenen 2 (n = 1,2), die Elektronenacceptor- und Allylsilan-Gruppen enthalten. Eine photochemische Cycloaddition liefert die 2-Hydroxytetrahydropyridine 3, die nach Zugabe von Säuren zu den Azabicyclen 4 cyclisieren. Hierbei greift in der Iminium-Ion-Zwischenstufe die Allylsilan-Gruppe C2 anti-ständig zum C3-gebundenen H-Atom an.
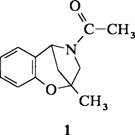
4-Acyl-2,3,4,5-tetrahydro-2-methyl-2,5-methano-benz-1,4-oxazepine, Derivate eines neuen Heterocyclus†
- Pages: 1667-1669
- First Published: Dezember 1991
Zwei Rotamere, die im festen Zustand bei Raumtemperatur stabil sind und getrennt isoliert werden konnten, bildet der Heterocyclus 1. Durch Variieren der Kristallisationsmethode konnten zwei Modifikationen erhalten werden, die sich in der Stellung der Carbonylgruppe am Stickstoffatom unterscheiden: Entweder zeigt die CO-Gruppe zum Benzolring (Bild rechts) oder sie weist davon weg.
Zweifache Insertion von Alkinen in Chrom-Kohlenstoff-Bindungen β-donorsubstituierter Vinylcarbenchrom-Komplexe - ein einfacher Zugang zu Cyclopenta[b]pyranen†‡
- Pages: 1669-1671
- First Published: Dezember 1991
Produkte mit stark eingeschränktem Substitutionsmuster, vielstufige Synthesen und niedrige Ausbeuten, das sind die Nachteile der bisher bekannten Synthesen für Pseudoazulene. Hier wird dagegen ein einfacher Zugang über die leicht erhältlichen Vinylcarbenchrom-Komplexe 1 beschrieben, die in guten Ausbeuten die Cyclopenta[b]pyrane 2 liefern, wenn in β-Position eine quartäre Gruppe gebunden ist. Die Röntgenstrukturanalyse zeigte, daß die beiden Ringe des Heterobicyclus 2 nur um 3.7° von der Planarität abweichen.
Synthese und Struktur des ersten Tellur(III)-Radikalkations†‡
- Pages: 1671-1672
- First Published: Dezember 1991
Monomeres Vorliegen im AsF6-Salz ergab die Tieftemperaturröntgenstrukturanalyse für das erste TeIII-Radikalkation 1. 1-AsF6 wurde überraschend bei der Umsetzung von Te(N(SiMe3)2)2 mit AgAsF6 in Form schwarzer Kristalle erhalten. Deren blaue, metastabile Lösungen in CH2Cl2 und CHCl3 zeigen nur einen einzigen breiten ESR-Peak, der darauf hinweist, daß sich das einzelne Elektron in einem p-Orbital am Telluratom befindet.
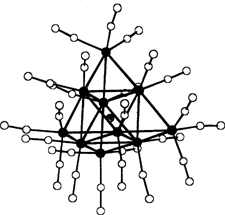
Synthese des ersten Nitridodecametall-Clusters; Kristallstrukturanalyse und 14N-NMR-Untersuchungen von [(Ph3P)2N][Ru10N(CO)24]†
- Pages: 1672-1674
- First Published: Dezember 1991
Ein vierfach überdachtes oktaedrisches Metallgerüst, das bisher nur von Carbido- und Hydrido-Clustern bekannt war, bildet das Monoanion [Ru10N(CO)24]− (Bild rechts). Dieser Cluster ist auf mehreren Wegen herzustellen; er entsteht sauber und ohne nicht abtrennbare Verunreinigungen bei der Umsetzung des oktaedrischen Clusters [Ru6N(CO)16]− mit [Ru3(CO)12].
Synthese und Struktur von Ethinylisocyanid†
- Pages: 1674-1676
- First Published: Dezember 1991
Unerwartet stabil ist Ethinylisocyanid 1, die Stammverbindung der bislang unbekannten Alkinylisocyanide. Es ist bei - 196°C mehrere Wochen und bei Raumtemperatur in der Gasphase einige Tage haltbar. Erhalten wurde 1 isomerenrein durch Blitz-Vakuumpyrolyse des Chromkomplexes 2. Die in dieser Arbeit beschriebenen Mikrowellendaten von 1 haben inzwischen bereits seine radioastronomische Identifizierung als Teil eines weiteren interstellaren, stickstoffhaltigen Isomerenpaares (mit HCC-CN) aus ungeladenen Molekülen mit gefüllten Elektronenschalen ermöglicht.
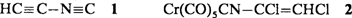
Diastereo- und enantioselektive, durch Michael-Additionen initiierte Cyclisierungen zu trans-substituierten Cyclopentancarbonsäureestern†‡
- Pages: 1676-1678
- First Published: Dezember 1991
Mit Diastereomeren- und Enantiomerenüberschüssen größer 95% können die Cyclopentancarbonsäureester 3 durch asymmetrische MIRC (Michael Initiated Ring Closure)-Reaktionen der Hydrazone 1 mit dem ungesättigten, bromierten Ester 2 hergestellt werden. Die de- und ee-Werte wurden gaschromatographisch sowie durch NMR-Experimente ermittelt. R = Me, nBu, iBu, Ph, p-BrC6H4.
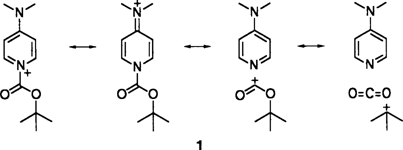
Kristallstrukturanalyse eines fragmentierbaren N-Acylpyridinium-Ions†
- Pages: 1678-1679
- First Published: Dezember 1991
Durch Konjugation im π- und im σ-System wird die positive Ladung im N-tert-Butyloxycarbonyl-4-dimethylaminopyridinium-Ion 1 stabilisiert. Dies folgt aus den röntgenographisch bestimmten Abständen und Winkeln im Salz 1-BF4. Die Strukturmerkmale sind auch in Einklang damit, daß 1 leicht Boc überträgt und daß Boc in CO2 und Isobuten zerfallen kann.
Tandem-Silylwanderungen zu α,β-bissilylierten Enalen und Enonen†‡
- Pages: 1679-1680
- First Published: Dezember 1991
Eine Reaktionssequenz aus einer Silyl-Wittig-Umlagerung und einer weiteren 1,2-Silylwanderung liefert die α,β-ungesättigten Ketone oder Aldehyde 3 aus den Alkinylethern 1. Als Zwischenprodukte entstehen die β-Alkinole 2, die trotz der vielen funktionellen Gruppen bei 0°C lagerfähig sind. 2 und 3 sind nützliche Synthesebausteine (R1 = Me, tBu, CMe-iPr; R2 = H, Me).
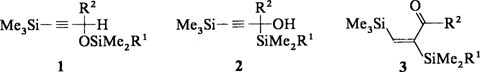
(NBu4)[Mn4O2(H2O)(O2 CPh)9], ein Butterfly-Komplex mit H2O als Ligand und sein Einsatz bei der Herstellung von acht- und elfkernigen Metallkomplexen†
- Pages: 1681-1683
- First Published: Dezember 1991
Für Modellsysteme für das Photosynthesezentrum der Wasseroxidation (WOC) bedeutet die Herstellung des Titelkomplexes (Bild rechts) einen großen Fortschritt, da bisher keine Modellverbindung ein gebundenes H2O-Molekül enthielt. Die Wasserstoffbrückenbindung des Aqualiganden (O 18) zu einer Carboxylatgruppe könnte auf die Art der Substratbindung im WOC sowie auf die Aktivierung zur Deprotonierung bei der H2O-Oxidation hinweisen.

Über den reaktionsbeschleunigenden Nachbargruppen-Effekt bei der Umsetzung von Vinylbromiden mit Alkylübergangsmetall-Reagentien†‡
- Pages: 1683-1685
- First Published: Dezember 1991
Nicht das Elektronendonorzentrum selbst, sondern vermutlich das in einer Zwischenstufe, z.B. 1, daran gebundene Metallatom verursacht den Nachbargruppeneffekt, der dazu führt, daß α- oder β-Donorsubstituenten (OH, OMe, NC) wie in 2 die alkylierende Substitution an Vinylbromiden mit Alkylübergangsmetall-Reagentien wie Me4MnLi2 stark beschleunigen. Auch die stärkere Beschleunigung durch α-Substituenten (n = 0) ist mit Zwischenstufen wie 1 in Einklang, da sich Fünfringe bekanntlich leichter bilden als Sechsringe. [M] = Fe-, Mn-Komplexfragmente.
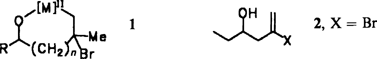
Neue chirale stationäre Polyamid-Phasen für die chromatographische Enantiomerentrennung†
- Pages: 1685-1687
- First Published: Dezember 1991
Die diastereomeren Polyamide 1 und 2 liefern stationäre Phasen für Enantiomerentrennungen mit sehr hoher Enantioselektivität. Diese Methacryloylamide sind auch im kg-Maßstab in guten Ausbeuten erhältlich und lassen sich sowohl auf stationäre Phasen aufpolymerisieren als auch mit einem Vernetzer zu Perlpolymerisaten für präparative Trennungen umsetzen.
Selektive para-Chlorierung von Biphenyl in L-Zeolithen†‡
- Pages: 1687-1689
- First Published: Dezember 1991
Ein industriell attraktiver Zugang zu 4,4′-Dichlorbiphenyl 1 ist die Chlorierung von Biphenyl mit Cl2 in Gegenwart metallausgetauschter L-Zeolithe. Die höchste Selektivität (96.7%) wurde in Dichlormethan bei 40°C mit Li-Zeolith L und einem 10proz. Überschuß an Cl2 erzielt. 1 ist ein interessantes Vorprodukt für Hochleistungspolymere, das möglichst ohne andere polychlorierte Biphenyle erhalten werden sollte.

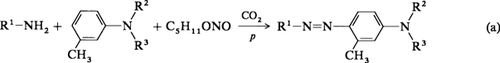
Salzfreie Synthese von Azo- und Hydrazonfarbstoffen unter CO2-Druck†
- Pages: 1689-1690
- First Published: Dezember 1991
Einen großen ökologischen Vorteil hat die Synthese von Azofarbstoffen gemäß Gleichung (a). Die salpetrige Säure wird hier aus ihren Salzen oder Estern mit CO2 unter Druck freigesetzt. Dies vermindert die Menge der als Nebenprodukte anfallenden Salze erheblich. In dieser einstufigen Reaktion können auch heterocyclische Amine erfolgreich diazotiert und mit tertiären aromatischen Aminen quantitativ gekuppelt werden (z.B. R1 = 3,4-Diazapyrrol-2-yl, R2 = Ethyl, R3 = Benzyl).
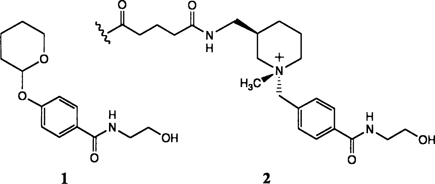
Antikörper-katalysierte Hydrolyse glycosidischer Bindungen†
- Pages: 1690-1692
- First Published: Dezember 1991
Auf dem Weg zu glycosidisch aktiven Abzymen ist man mit der Generierung von katalytischen Antikörpern, die die Hydrolyse des Acetals 1 beschleunigen, einen guten Schritt vorangekommen. Die Antikörper (= Abzyme) wurden durch Immunisierung mit dem Piperidinium-Hapten 2 erhalten. Entscheidend für die katalytische Aktivität dieses neuen Abzyms scheint zu sein, daß die Abgangsgruppe von 1 in der Antikörperbindungstasche in eine axiale Stellung gezwungen wird.
Metallorganische Chemie an Oxidoberflächen: Selektive, katalytische Tieftemperatur-Hydrogenolyse von Alkanen durch ein sehr elektrophiles Zirconiumhydrid auf Kieselgel†
- Pages: 1692-1694
- First Published: Dezember 1991
Bereits bei 50 °C katalytisch wirksam ist auf Kieselgel aufgebrachtes Zirconiumhydrid [(
Ein neues chromogenes β-Galactosidase-Substrat: 7-β-D-Galactopyranosyloxy-9,9-dimethyl-9H-acridin-2-on†‡
- Pages: 1694-1696
- First Published: Dezember 1991
Eine Farbverschiebung um 196 nm (438 → 634 nm) zeigt die Titelverbindung 1 bei Hydrolyse bei pH = 8. Die Totalsynthese, die kinetischen Parameter und die Anwendung von 1 bei Enzym-Immunoassays werden beschrieben.
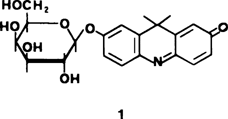
Die Reaktion von PtCl2 mit einem N,S,O-koordinierten Calciumkomplex: Synthese und Struktur eines Ca-Pt-Komplexanions†
- Pages: 1696-1697
- First Published: Dezember 1991
Aus einem Ca2-Komplexkation und einem Ca-Pt-Komplexanion (siehe Bild rechts) besteht das Salz 1, das bei der Reaktion von [(Ox)2Ca(HMPA)2] mit PtCl2 in siedendem Toluol gebildet wird. Das Ca-Pt-Komplexanion ist als Modellverbindung für heteronucleare Katalysatoren und für Addukte von Pt2+ mit DNA-Basen von Interesse.
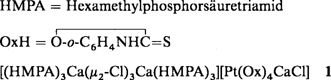

Neuartige vierkernige Hexapentaen-Eisenkomplexe: Erste Beispiele von Poly-π-allylkomplexen
- Pages: 1697-1699
- First Published: Dezember 1991
Vier Fe(CO)3-Gruppen an eine C6-Kette π-allylartig koordiniert, so lassen sich die Komplexe 2 und 3 beschreiben, die bei der Umsetzung von Hexapentaen 1 (R = H) und 2,7-Dimethylocta-2,3,4,5,6-pentaen 1 (R = CH3) mit dem Komplex [Fe3(CO)12] in geringer Ausbeute entstehen. Strukturbestimmungen und die theoretische Analyse der Bindungsverhältnisse ergaben übereinstimmende Ergebnisse, z.B. daß die zentrale C-C-Bindung durch die Koordination am stärksten geschwächt (und gedehnt) wird.
Diastereomerentrennung durch Bildung von Einschlußverbindungen: 2,6-Dimethylnaphthalin-Komplexe von Flumethrin†
- Pages: 1699-1701
- First Published: Dezember 1991
Das insektizid und akarizid unwirksame Flumethrin-Diastereomer läßt sich aus dem technischen Isomerengemisch durch die Bildung von Einschlußverbindungen vom Typ 1 abtrennen, wodurch der Anteil an wirksamem Diastereomer größer und somit die Aufwandmenge an Insektizid kleiner wird.
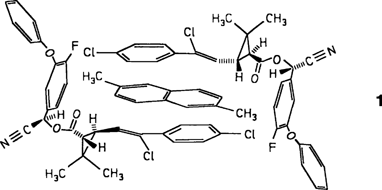
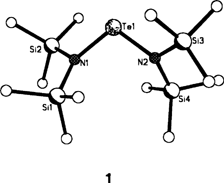
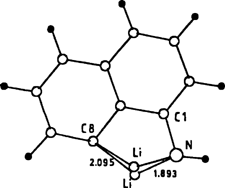
Dilithiierung eines primären Amins: Synthese und Struktur von [(α-Naphthyl-NLi2)10(Et2O)6] · Et2O, einem paramagnetischen N10Li20-Aggregat†
- Pages: 1702-1704
- First Published: Dezember 1991
Am stabilsten ist nach ab-initio-Rechnungen das rechts abgebildete Isomer von dilithiiertem α-Naphthylamin, bei dem C8 und N deprotoniert sind und die beiden Li-Atome eine Brückenposition zwischen diesen Atomen einnehmen. Die Umsetzung von α-Naphthylamin mit nBuLi bei - 40°C in Diethylether ergibt den Titelkomplex, dessen Struktur durch einen zentralen N10Li14-Cluster gekennzeichnet ist, der durch zwei rhombische Dodecaeder mit einer gemeinsamen Fläche gebildet wird. Erstaunlicherweise ist die Verbindung in Lösung und im Festkörper paramagnetisch.
Methyltrioxorhenium als Katalysator für die Olefin-Metathese†‡
- Pages: 1704-1706
- First Published: Dezember 1991
Ohne Zusatz von Cokatalysatoren katalysiert Methyltrioxorhenium (MTO) die Metathese funktionalisierter Olefine, wenn es auf sauren Trägermaterialien eingesetzt wird. Typisch ist das System MTO/Al2O3-SiO2, das unter anderem die Methathese von Allylhalogeniden, Allylsilanen, ungesättigten Carbonsäureestern und Nitrilen bewerkstelligt. Als Homogenkatalysator ist MTO in der Kombination MTO/RnAlCl3-n in Ringöffnungspolymerisationen aktiv (R = CH3, C2H5; n = 1, 2).
Methyltrioxorhenium als Katalysator für die Olefin-Oxidation†‡
- Pages: 1706-1709
- First Published: Dezember 1991
Ausgeprägte Lewis-Acidität ist die Erklärung für die außergewöhnliche Aktivität der Titelverbindung in der katalytischen Olefin-Epoxidierung mit Wasserstoffperoxid. Das System MTO/t-C4H9OH/H2O2 epoxidiert selektiv und effizient Olefine unter sehr schonenden Bedingungen (- 30 bis + 60°C, in der Regel Raumtemperatur). 0.1 bis 1 Mol-% MTO sind im allgemeinen zur Erreichung hoher Umsätze ausreichend. Durch Variation der Alkylgruppe sowie durch die Zugabe von Cokatalysatoren (z.B. Aminen) sollten sich Aktivität und Stereoselektivität beeinflussen lassen.
Methyltrioxorhenium als Katalysator einer neuen Aldehyd-Olefinierung†‡
- Pages: 1709-1711
- First Published: Dezember 1991
Aus Aldehyden oder cyclischen Ketonen, Diazoalkanen und tertiären Phosphanen lassen sich nach Gleichung (a) MTO-katalysiert Olefine synthetisieren. Insbesondere Diazoacetate und -malonate (R2, R3 = H, CO2Et bzw. 2 x CO2Me) lassen sich so mit aliphatischen und aromatischen Aldehyden (R1 = iPr, trans-PhCH = CH, Ph, 4-NO2C6H4 etc.) zu Olefinen umsetzen. Leicht zugängliche Startverbindungen, einfache Durchführbarkeit, milde Reaktionsbedingungen und gute Ausbeuten charakterisieren die neue Synthesemethode (R′ = Ph, 3-C6H4SO3Na, nBu).

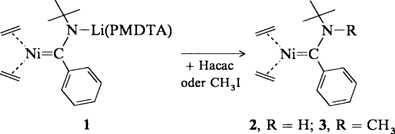
Nickel(0)-Carbenkomplexe†
- Pages: 1711-1713
- First Published: Dezember 1991
Ein Tetracarbennickel(0)-Komplex konnte noch nicht synthetisiert werden, doch ergaben Studien zu diesem Thema andere interessante Resultate. So entstehen die 16e-Nickel(0)-Carbenkomplexe 2 und 3 bei der Protonierung bzw. Methylierung des aus Tris(ethen)nickel(0) und dem Additionsprodukt von Phenyllithium an tert-Butylisocyanid gewonnenen Komplexes 1 (PMDTA = Pentamethyldiethylentriamin).
Tetraalkoxytitan-Carbenkomplexe mit zweifacher intramolekularer Et2Al-Verbrückung†
- Pages: 1714-1715
- First Published: Dezember 1991
Der erste sich nicht von Titanocen ableitende Titan-Carbenkomplex 1 bildet sich bei der Umsetzung eines Titan(IV)-diolats mit AlEt3. Die Struktur dieses Heterobimetall-Carbenkomplexes erinnert stark an die der Tebbe-Reagentien. 3,3-Dimethylcyclopropen reagiert mit 1 unter Ringöffnung und liefert einen analogen Carbenkomplex.

Röntgenkleinwinkelanalyse von polymeren Latices mit Kern-Schale-Morphologie†
- Pages: 1715-1717
- First Published: Dezember 1991
Den inneren Aufbau von Kern-Schale-Latices zu analysieren, ohne diese vorbehandeln zu müssen, ermöglicht die Röntgenkleinwinkelstreuung in Kombination mit der Ultrazentrifugenanalyse. Letztere wird dabei zur Bestimmung der Teilchengrößenverteilung herangezogen. Für größere Streuwinkel (q > 0.1 nm−1) erhält man eine quantitative Übereinstimmung von gemessenen und berechneten Streuintensitäten; die Abweichungen im Bereich kleinster q-Werte sind auf interpartikuläre Interferenzen zurückzuführen und geben Aufschluß über die Wechselwirkung der Teilchen.
Drei- und vierfach brückenkopfsubstituierte Tribenzotriquinacene†‡
- Pages: 1717-1720
- First Published: Dezember 1991
Schlüsselzwischenverbindungen für die Synthese brückenkopfsubstituierter Tribenzotriquinacene ist das pyramidalisierte Olefin 1, das bis zu vierfach substituierte Tribenzotriquinacene ebenso zugänglich machte wie das unsubstituierte Cyclopropatribenzotriquinacen.
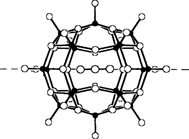
Templatgesteuerte Bildung von Clusterschalen oder eine Art molekulare Erkennung: Synthese von [HV22O54(ClO4)]6− und [H2V18O44(N3)]5−†
- Pages: 1720-1722
- First Published: Dezember 1991
Anionen als Gäste in Anionen bestimmen als Template die Form molekularer Hohlräume (Clusterschalen), die durch verschiedene Verknüpfungen einer tetragonal-pyramidalen OVO4-Basiseinheit entstehen. Im Falle der Cluster-Anionen [HV22O54(ClO4)]6− und [H2V18O44(N3)]5− (Strukturbild rechts) z.B. erfolgt der Aufbau der Clusterschalen entsprechend einer „molekularen”︁ Erkennung der (O-O)2- bzw. N(N)N-Einheiten.
Neue Wege zur Synthese Nitrilgruppen enthaltender Polymere†‡
- Pages: 1722-1725
- First Published: Dezember 1991
Polymere, die aus reinen CC-Hauptketten und Nitrilsubstituenten bestehen, versprechen, leistungsfähige Elastomere zu sein. Dieser Polymertyp konnte nun auf zwei Wegen hergestellt werden: einerseits durch direkte Excimerlaser-induzierte radikalische Copolymerisation von Ethen und Acrylnitril, andererseits durch polymeranaloge Umsetzung eines Ethen/Acrylat-Copolymers in verdichtetem fluiden Ammoniak [Gl.(a)].
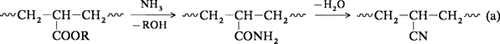
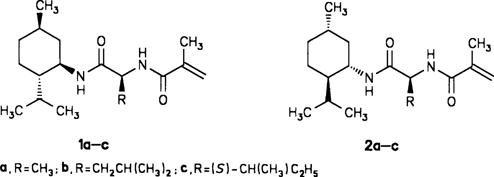
Stereoselektive Synthese von cyclischen Hydroxyalkyl-Dipeptidisosteren über metallierte N,N-Dialkylcarbamidsäure-2-alkenylester†‡
- Pages: 1725-1727
- First Published: Dezember 1991
Potentielle Proteaseinhibitoren entstehen stereoselektiv am Ende einer Reaktionssequenz, die mit der Umsetzung N-geschützter α-Aminoaldehyde mit dem Homoenolat-Reagens 1 beginnt. Die dabei gebildeten Lactone 2 lassen sich durch eine neue Variante der Weinreb-Methode, bei der Dialkylaluminiumamide als ringöffnende Reagentien eingesetzt werden, mit α-Aminosäureamiden kuppeln. So entstehen vier Diastereomere der Verbindungen 3, die zur Ψ-Phe-[CHOHCH]Pro-Gruppe gehören. R CF3CO, Cb C(CO)NiPr2, Bn PhCH2.
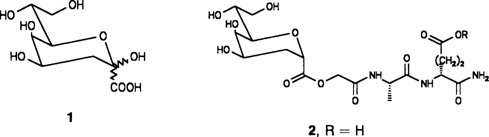
Synthese von Zellwandpeptidkonjugaten der 2,3-Didesoxy-β-D-manno-2-octulosonsäure†
- Pages: 1728-1729
- First Published: Dezember 1991
Ein Inhibitor der bakteriellen Lipopolysaccharid-Biosynthese ist die Titelverbindung 2-Desoxy-β-KDO 1, die allerdings die Bakterienmembran nicht durchdringen kann. Die Verknüpfung der Carboxyfunktion mit peptidischen Carriern führt zu Konjugaten (Prodrugs) wie 2, deren in-vivo-Stabilität durch Modifizierung der potentiellen Spaltstellen moduliert werden kann.
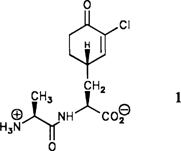
Enantio- und diastereoselektive Totalsynthese des antimykotisch wirkenden Naturstoffes Chlorotetain: Revision der relativen Konfiguration†‡
- Pages: 1729-1731
- First Published: Dezember 1991
Mit der Bislactimether-Methode und durch Deprotonierung mit einer chiralen Base gelang der enantio- und diastereoselektive Aufbau der ungewöhnlichen C-terminalen Aminosäure des Dipeptids Chlorotetain 1, eines Naturstoffes aus Bacillus subtilis. Chlorotetain 1 ist, entgegen der Literaturangabe, im Cyclohexenylrest der Seitenkette (S)-konfiguriert.
Synthese eines elicitoraktiven Heptaglucansaccharides zur Untersuchung pflanzlicher Abwehrmechanismen†
- Pages: 1731-1732
- First Published: Dezember 1991
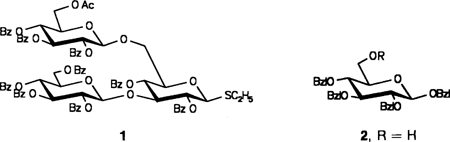
Geringe Stufenzahl und sehr gute Ausbeuten zeichnen die Blocksynthese eines Heptasaccharids aus, das die kleinste stark elicitoraktive Einheit im Wirt-Parasit-System Sojabohne/Phytophthora megasperma ist, die an einen Rezeptor bindet. Zentrale Zwischenstufen der Blocksynthese sind der Thiotrisacchariddonor 1 und der Glycosylacceptor 2 (Ac = CH3CO, Bz = C6H5CO, Bzl = C6H5CH2).
Tetrakis(dimethylamino)ethen: Ein extrem elektronenreiches Molekül mit ungewöhnlicher Struktur sowohl im Festkörper als auch in der Gasphase†‡
- Pages: 1733-1735
- First Published: Dezember 1991
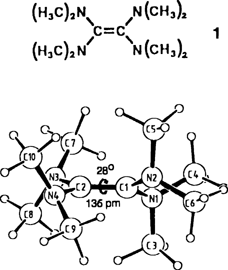
Ein ungewöhnliches und chirales Molekülgerüst charakterisiert die bei 273 K erstarrende, luftempfindliche Titelverbindung 1 im Festkörper. Die Molekülhälften beiderseits der auf 138 pm gestreckten CC-Achse sind um 28° gegeneinander verdrillt (Bild rechts). Auch in der Gasphase weist 1 eine durch sterische Überfüllung bedingte ungewöhnliche Molekülstruktur auf, in der die vier Stickstoff-Elektronenpaare der erheblich eingeebneten Dimethylaminosubstituenten mit Torsionswinkeln ω(CC-NC2) von 55° aus den optimalen π-Wechselwirkungspositionen ausgelenkt sind.
1,3-Dipolare Cycloaddition als Schlüsselreaktion für die Synthese potenter Renin-Inhibitoren†
- Pages: 1735-1737
- First Published: Dezember 1991
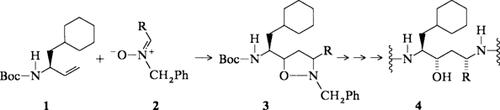
Substituierte Allylamine wie 1 reagieren mit Nitronen wie 2 unter 1,3-Dipolarer Cycloaddition zu diastereomeren Isoxazolidinen wie 3, die chromatographisch getrennt werden können. Hydrogenolytische Öffnung der Isoxazolidine ergibt 1,3-Aminoalkohole, die - in die Angiotensinogen-Sequenz eingebaut - zu Renin-Inhibitoren vom Typ 4 führen.
Isophlorine: Moleküle am Schnittpunkt von Porphyrin- und Annulen-Chemie†‡
- Pages: 1737-1741
- First Published: Dezember 1991
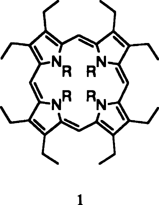
Die bereits von Woodward bei der Chlorophyll-Synthese diskutierten Isophlorine sind als janusköpfige Moleküle - einerseits Porphyrine (N,N′-Dihydroporphyrine), andererseits echte Annulene (der Hückel-Regel nicht entsprechende [20]Annulene) - strukturchemisch von besonderem Reiz. Während NH-unsubstituierte Isophlorine nach wie vor der Entdeckung oder Synthese harren, konnte das N,N′,N″,N″'-Tetramethyloctaethylisophlorin 1, R CH3, jetzt durch Zweielektronen-Reduktion des Franckschen N,N′,N″,N″'-Tetramethyloctaethylporphyrin-Dikations gewonnen werden.
Ein neuer Zugang zu Zweiäquivalentkupplern für die Farbphotographie†
- Pages: 1742-1743
- First Published: Dezember 1991
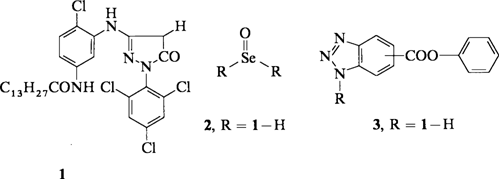
Durch Einführung der Selen(IV)-Gruppe in die Kupplungsstelle von Anilinopyrazolonen und Naphtholen, z.B. mit SeO2, gelingt erstmals die direkte Umwandlung der entsprechenden Vieräquivalentkuppler in Zweiäquivalentkuppler mit einer Triazolfluchtgruppe. So kann 1 mit SeO2 zu 2 umgesetzt werden, das mit einem Benzotriazol unter Selenabspaltung zu 3 reagiert.
Sign up for email alerts
Tools
Eine Zeitschrift der
Gesellschaft Deutscher Chemiker
More from this journal
Classifieds: Jobs and Awards, Products and Services
- Angewandte Chemie
- 4 November 2024
Graphical Abstract

Are you looking for a (new) job? Do you know someone who deserves an award? Are you trying to find a product or service to make your work more efficient? Our partners can help.
Related Titles



 IssueVolume 97, Issue 7
IssueVolume 97, Issue 7Special Issue: Membrantechnik
July 2025Guest Editor(s):
Deutsche Gesellschaft für Membrantechnik