

Journal list menu

 Issue2019i, 1623-1904
Issue2019i, 1623-1904
Export Citations
Download PDFs
Front Cover, Volume 40, Issue 10
- Page: i
- First Published: 27 September 2019

Front Cover: The cover image is based on the RESEARCH ARTICLE Mutations in GDF11 and the extracellular antagonist, Follistatin, as a likely cause of Mendelian forms of orofacial cletiing in humans by Timothy C. Cox et al., https://doi.org/10.1002/humu.23793.
Cover image © Timothy Cox Images.
Issue Information
- Pages: 1623-1626
- First Published: 27 September 2019
In Memoriam: Emeritus Professor Sue (Margaret Susan) Povey [1942–2019]
- Pages: 1627-1629
- First Published: 25 July 2019
Phenotype predictions for exon deletions/duplications: A user guide for professionals and clinicians using Becker and Duchenne muscular dystrophy as examples
- Pages: 1630-1633
- First Published: 29 July 2019
Checklist for gene/disease-specific variation database curators to enable ethical data management
- Pages: 1634-1640
- First Published: 26 July 2019
Mutation update on ACAT1 variants associated with mitochondrial acetoacetyl-CoA thiolase (T2) deficiency
- Pages: 1641-1663
- First Published: 03 July 2019
A Vietnamese human genetic variation database
- Pages: 1664-1675
- First Published: 10 June 2019
The novel p.Ser263Phe mutation in the human high-affinity choline transporter 1 (CHT1/SLC5A7) causes a lethal form of fetal akinesia syndrome
- Pages: 1676-1683
- First Published: 12 July 2019
Genome-wide DNA methylation and RNA analyses enable reclassification of two variants of uncertain significance in a patient with clinical Kabuki syndrome
- Pages: 1684-1689
- First Published: 03 July 2019
Three new cases of ataxia-telangiectasia-like disorder: No impairment of the ATM pathway, but S-phase checkpoint defect
- Pages: 1690-1699
- First Published: 29 April 2019
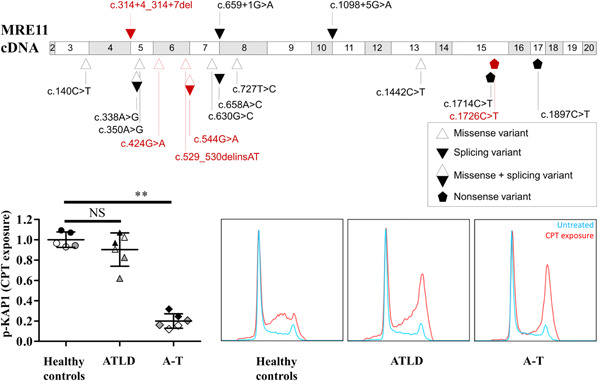
Ataxia-Telangiectasia-Like Disorder (ATLD) is a rare genomic instability syndrome caused by biallelic variants of MRE11 (meiotic recombination 11) characterized by progressive cerebellar ataxia. We describe the first three French patients with progressive cerebellar ataxia diagnosed with ATLD, associated with compound heterozygosity for unreported MRE11 variants. Functional consequences were evaluated on activation of the ataxia-telangiectasia mutated (ATM) pathway via phosphorylation of ATM targets, but no consistent defect was observed. However, an S-phase checkpoint activation defect after camptothecin was observed in these ATLD patients.
Mutations in TIMM50 cause severe mitochondrial dysfunction by targeting key aspects of mitochondrial physiology
- Pages: 1700-1712
- First Published: 06 May 2019
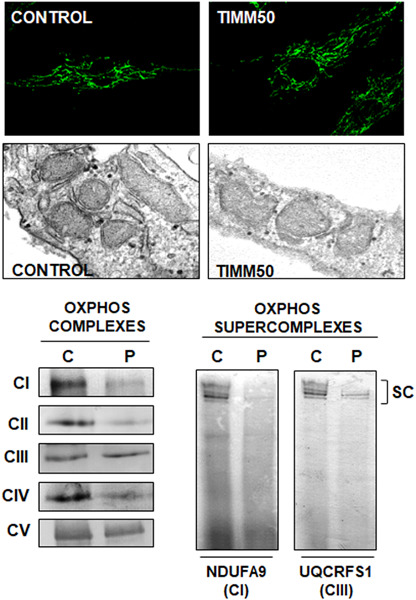
TIMM50 deficiency targets key aspects of mitochondrial physiology, such as the maintenance of mitochondrial morphology, OXPHOS complexes and supercomplexes assembly, and mitochondrial respiratory capacity.
Functional classification of ATM variants in ataxia-telangiectasia patients
- Pages: 1713-1730
- First Published: 03 May 2019

Ataxia-telangiectasia is a recessive disorder caused by biallelic pathogenic variants of ataxia-telangiectasia mutated. This disease is characterized by progressive ataxia, telangiectasia, immune deficiency, predisposition to malignancies, and radiosensitivity.
Mutations in ELAC2 associated with hypertrophic cardiomyopathy impair mitochondrial tRNA 3′-end processing
- Pages: 1731-1748
- First Published: 02 May 2019
Cost-effective molecular inversion probe-based ABCA4 sequencing reveals deep-intronic variants in Stargardt disease
- Pages: 1749-1759
- First Published: 18 June 2019
Neurofibromatosis type 1-related pseudarthrosis: Beyond the pseudarthrosis site
- Pages: 1760-1767
- First Published: 08 May 2019
A novel mutation in the erythroid transcription factor KLF1 is likely responsible for ameliorating β-thalassemia major
- Pages: 1768-1780
- First Published: 22 May 2019
BRCA1 and BRCA2 pathogenic sequence variants in women of African origin or ancestry
- Pages: 1781-1796
- First Published: 21 May 2019
Increasing phenotypic annotation improves the diagnostic rate of exome sequencing in a rare neuromuscular disorder
- Pages: 1797-1812
- First Published: 23 June 2019
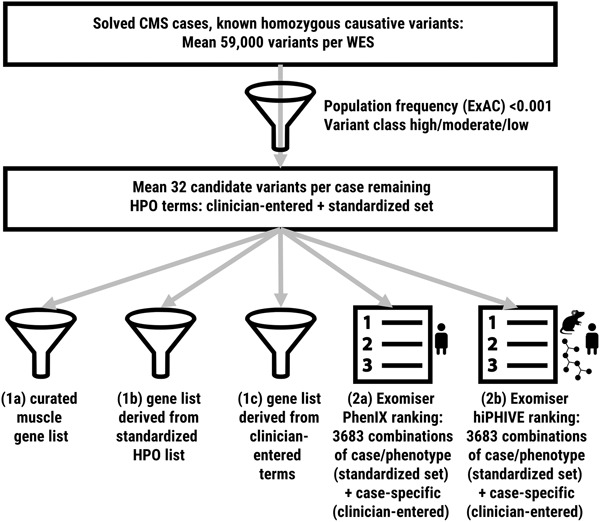
In this study, we use a cohort of solved congenital myasthenic syndrome cases to test methods for variant interpretation with the aid of human phenotype ontology annotations. We show that gene-list filters created from phenotypic annotations perform similarly to curated disease-gene virtual panels, that Exomiser is a valuable aid to variant discovery, and that annotation with over five terms is recommended in our context.
Mutations in GDF11 and the extracellular antagonist, Follistatin, as a likely cause of Mendelian forms of orofacial clefting in humans
- Pages: 1813-1825
- First Published: 18 June 2019
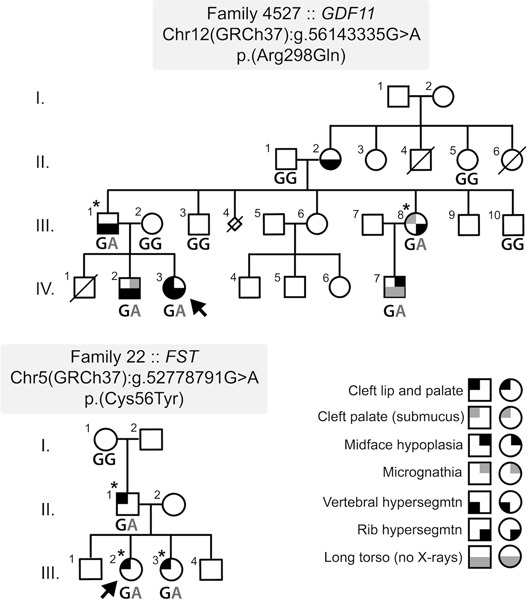
The authors report the identification, via exome sequencing, of segregating variants in two genes, GDF11 and FST (encoding the GDF11 antagonist, Follistatin), in families with cleft lip/palate. The GDF11 variant segregated with both cleft lip/palate, as well as rib and vertebral hypersegmentation, mirroring the Gdf11 knockout mouse, and suggesting a new clefting syndrome, whereas the family harboring the segregating FST variant exhibited isolated cleft lip/palate. Functional assays, together with biochemical, genetic, and expression data support the pathogenicity of the variants and highlight a previously unappreciated role for this pathway in human orofacial development.
Mutations in KARS cause a severe neurological and neurosensory disease with optic neuropathy
- Pages: 1826-1840
- First Published: 22 May 2019
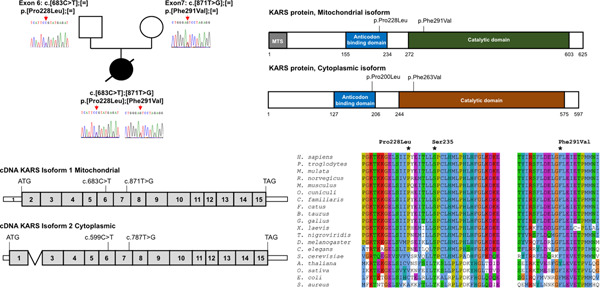
Here we report on a patient with severe neurological disease, found to carry biallelic mutations c.683C>T (p.Pro228Leu) and c.871T>G (p.Phe291Val) in the lysyl-tRNA synthetase KARS
Disease-associated missense variants in ZBTB18 disrupt DNA binding and impair the development of neurons within the embryonic cerebral cortex
- Pages: 1841-1855
- First Published: 21 May 2019

Here, we studied the zinc-finger repressor protein ZBTB18 and its disease-associated missense variants reported in subjects diagnosed with microcephaly (N461S, in yellow) and macrocephaly (R495G, in blue). We report that each of these missense ZBTB18 variants (i) influences the DNA-binding specificity of ZBTB18; (ii) its capacity for transcriptional regulation; as well as (iii) its neurodevelopmental signaling activity during cerebral cortex development. Our results suggest that altered transcriptional regulation could represent an important underlying pathological mechanism for missense ZBTB18 variants in cases of brain developmental disorder.
First estimate of the scale of canonical 5′ splice site GT>GC variants capable of generating wild-type transcripts
- Pages: 1856-1873
- First Published: 27 May 2019
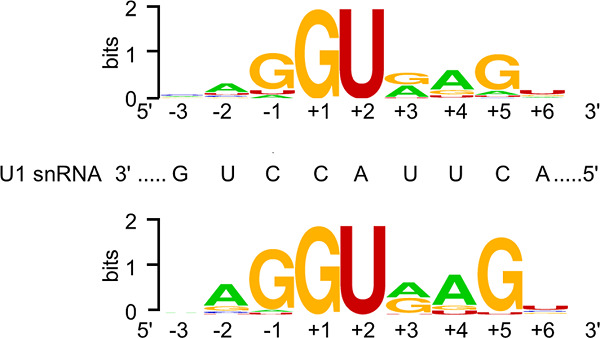
Based upon complementary data from the meta-analysis of 45 disease-causing 5′SS GT>GC variants and the cell culture-based full-length gene splicing analysis of 103 5′SS GT>GC substitutions, we have provided a first estimate of ~15–18% for the proportion of canonical GT 5′SSs that are capable of generating between 1% and 84% normal transcripts in case of the substitution of GT by GC. Given that even the retention of 5% normal transcripts can significantly ameliorate a patient's clinical phenotype, our findings imply the potential existence of hundreds or even thousands of disease-causing 5′SS GT>GC variants that may underlie relatively mild clinical phenotypes. As 5′SS GT>GC variants can also give rise to relatively high levels of wild-type transcripts, our findings imply that 5′SS GT>GC variants may not invariably be pathogenic in disease-causative or disease-associated genes.
Functional and cellular localization diversity associated with Fukutin-related protein patient genetic variants
- Pages: 1874-1885
- First Published: 03 July 2019
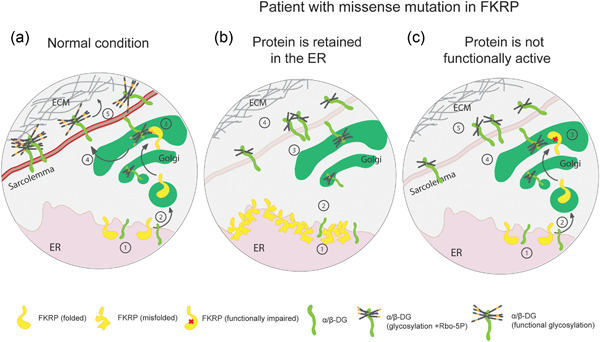
Graphical model illustrating the proposed fate of normal and abnormal FKRP proteins.
TAB2 c.1398dup variant leads to haploinsufficiency and impairs extracellular matrix homeostasis
- Pages: 1886-1898
- First Published: 27 June 2019
A mutation creating an upstream translation initiation codon in SLC22A5 5′UTR is a frequent cause of primary carnitine deficiency
- Pages: 1899-1904
- First Published: 12 June 2019