

Journal list menu

 Issue2013v, 1449-1587
Issue2013v, 1449-1587
Export Citations
Download PDFs
Loss-of-function Mutation in the NOTCH3 Gene: Simply a Polymorphism?
- Page: v
- First Published: 09 October 2013
The TREAT-NMD Duchenne Muscular Dystrophy Registries: Conception, Design, and Utilization by Industry and Academia
- Pages: 1449-1457
- First Published: 02 August 2013

TREAT-NMD is a worldwide network for neuromuscular diseases that provides an infrastructure to support the delivery of promising new therapies for patients.
The Finnish Disease Heritage Database (FinDis) Update—A Database for the Genes Mutated in the Finnish Disease Heritage Brought to the Next-Generation Sequencing Era
- Pages: 1458-1466
- First Published: 31 July 2013

The Finnish disease heritage refers to a group of rare monogenic diseases that are, by definition, more prevalent in Finland than elsewhere in the world. In the work data of the 36 diseases were updated and made available from a web portal (http://findis.org) based on LOVD instances, which are provided as a service for curators by the Leiden University Medical Center.
Novel FOXF1 Deep Intronic Deletion Causes Lethal Lung Developmental Disorder, Alveolar Capillary Dysplasia with Misalignment of Pulmonary Veins
- Pages: 1467-1471
- First Published: 13 August 2013
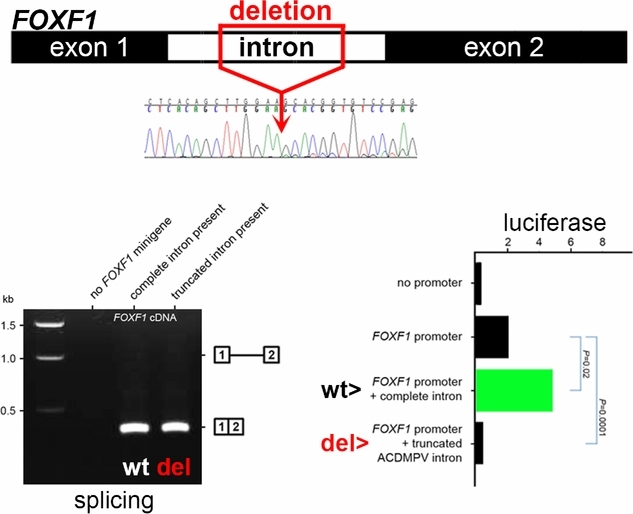
Haploinsufficiency of FOXF1 causes neonatal lethal Alveolar Capillary Dysplasia with Misalignment of Pulmonary Veins (ACDMPV). We report a novel pathogenic FOXF1 deep intronic deletion that did not affect major splicing sites. Results of the in vitro minigene-based splicing and reporter assays performed in normal fetal lung fibroblasts suggest that the deletion likely compromised the intronic element(s) apparently regulating FOXF1 transcription. Our data further emphasize the importance of inclusion of non-coding regions of the human genome in diagnostic testing.
Mutation in TTI2 Reveals a Role for Triple T Complex in Human Brain Development
- Pages: 1472-1476
- First Published: 16 August 2013
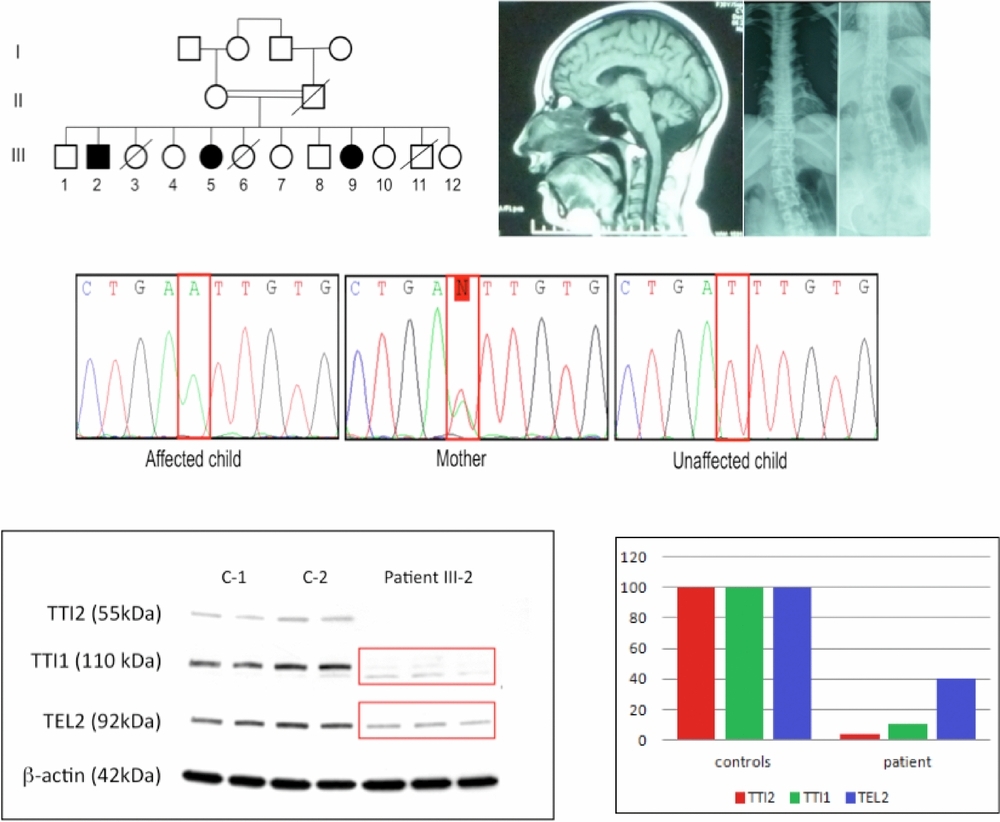
We identified a mutation in TTI2 as a novel cause of autosomal recessive syndromic intellectual disability. TTI2 encodes one of the subunits of the Triple T complex, a regulator of phosphoinositide-3-kinase-related protein kinase (PIKKs) abundance. We demonstrated impaired stability of the Triple T complex and reduced steady-state level of all PIKKs in patient cells. These findings assess the role of TTI2 in the aetiology of intellectual disability and further support the role of PIKK signalling in brain development and functioning.
Inactivation of DNA Mismatch Repair by Variants of Uncertain Significance in the PMS2 Gene
- Pages: 1477-1480
- First Published: 20 August 2013
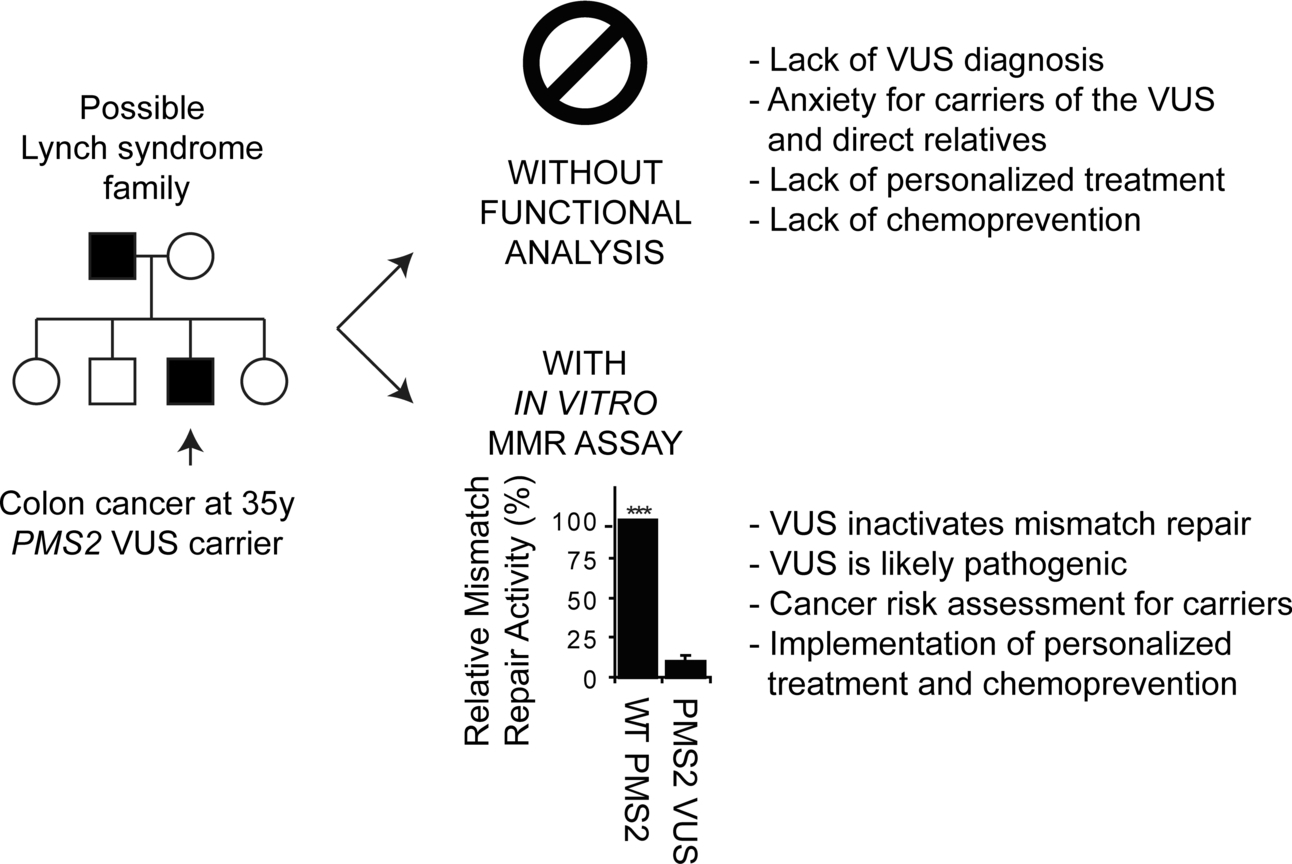
The cancer predisposition Lynch syndrome is diagnosed by the identification of a disruptive mutation in one of the DNA mismatch repair genes. Unfortunately, the identification of a variant of uncertain significance frequently precludes diagnosis, which interferes with clinical management. Here we use a rapid and cell-free assay to test functional activity of nearly all variants identified to date in PMS2, revealing loss of activity for five variants. This assay may greatly facilitate the diagnostic assessment of PMS2 variants.
Telomere Phenotypes in Females with Heterozygous Mutations in the Dyskeratosis Congenita 1 (DKC1) Gene
- Pages: 1481-1485
- First Published: 14 August 2013
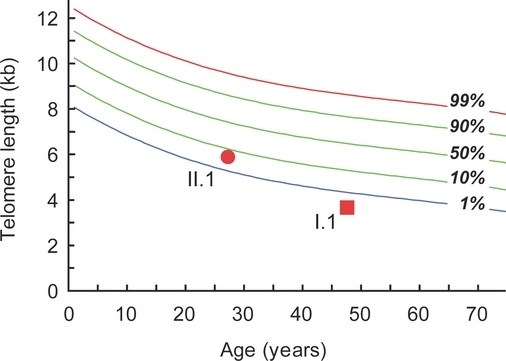
In this manuscript, we provide evidence that females with heterozygous mutations in the X-linked gene, DKC1, may at times be at risk for developing telomere-related phenotypes. These phenotypes appear in parenchymal organs such as the skin, and not in the blood, because of the competitive advantage of telomerase competent cells in the bone marrow.
Hypomorphic NOTCH3 Alleles Do Not Cause CADASIL in Humans
- Pages: 1486-1489
- First Published: 02 September 2013
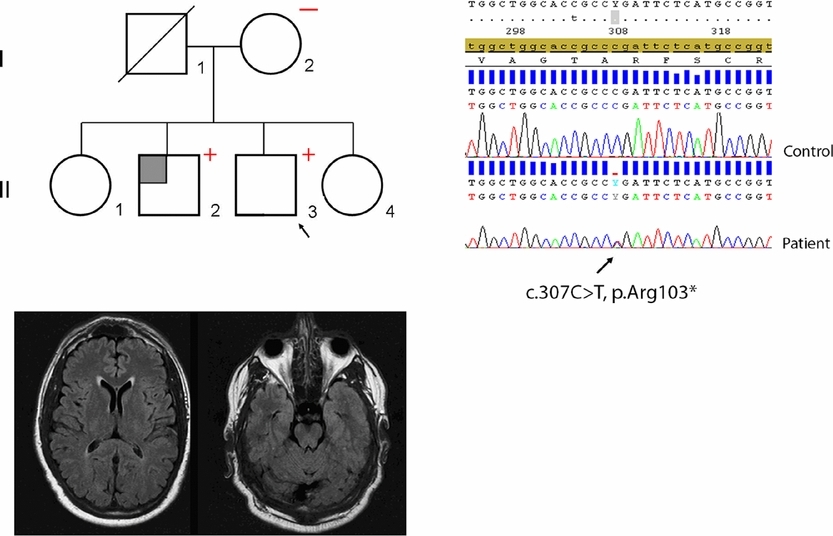
In this study, we address the previously unresolved issue of the role of hypomorphic NOTCH3 alleles in CADASIL. Based on extensive investigations of two families, we show that hypomorphic NOTCH3 alleles do not cause CADASIL. Furthermore, we describe the first patient who is compound heterozygous for a typical cysteine altering NOTCH3 missense mutation and a large intragenic NOTCH3 deletion who, interestingly, has a phenotype within the normal CADASIL spectrum.
In Vitro Secretion Deficits are Common Among Human Coagulation Factor XIII Subunit B Missense Mutants: Correlations with Patient Phenotypes and Molecular Models
- Pages: 1490-1500
- First Published: 02 August 2013
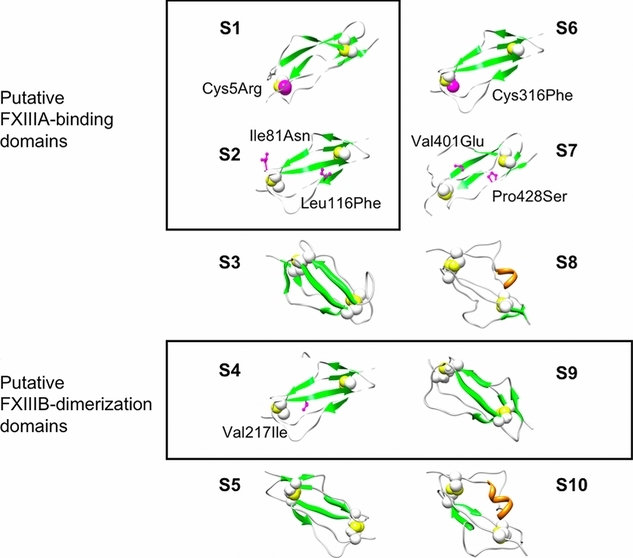
Pairs of coagulation factor XIII (FXIII) B subunits bound to A dimers prior to activation are thought to possess carrier/protective function. Mutations in either A or B subunits are associated with pathological patient phenotypes of mild to severe bleeding. HEK293T cell expression studies of seven recently identified FXIIIB missense variants revealed impaired in vitro secretion phenotypes. Molecular models of FXIIIB Sushi domains constructed using phylogenetically similar complement factor H Sushi domain templates suggest possible pathological mechanisms.
MTO1 Mutations are Associated with Hypertrophic Cardiomyopathy and Lactic Acidosis and Cause Respiratory Chain Deficiency in Humans and Yeast
- Pages: 1501-1509
- First Published: 08 August 2013
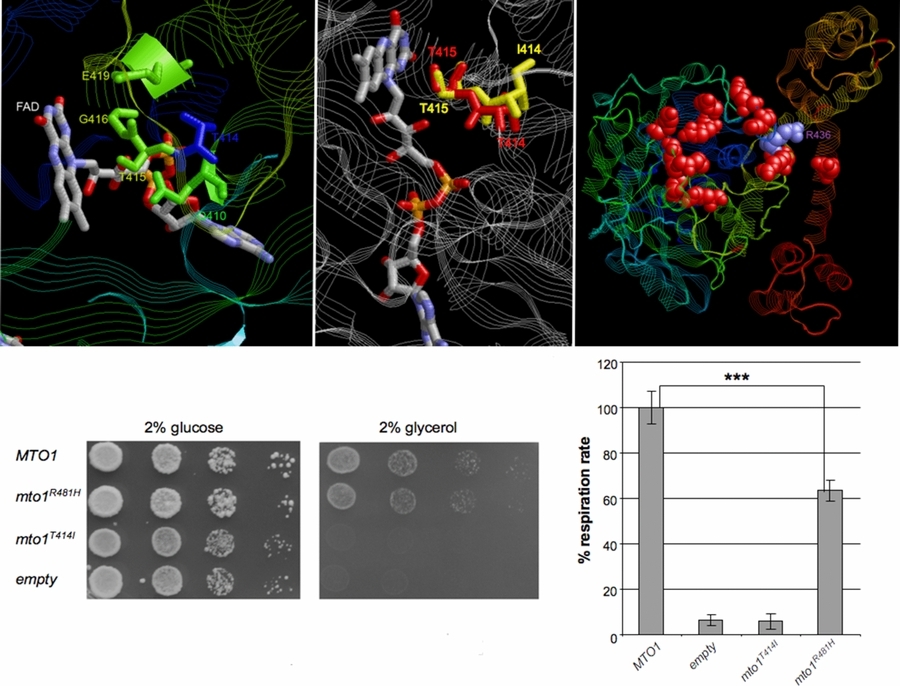
MTO1 mutations cause a mitochondrial disorder, characterized by hypertrophic cardiomyopathy, lactic acidosis, and mitochondrial respiratory chain deficiency. The pharmacological treatment, centered on the control of lactic acidosis by dichloroacetate, could have beneficial effects on clinical presentation and progression of the disease. A suitable recombinant yeast model can easily validate the pathogenic role of variants found in human patients with mitochondrial disorders.
NF1 Molecular Characterization and Neurofibromatosis Type I Genotype–Phenotype Correlation: The French Experience
- Pages: 1510-1518
- First Published: 02 August 2013
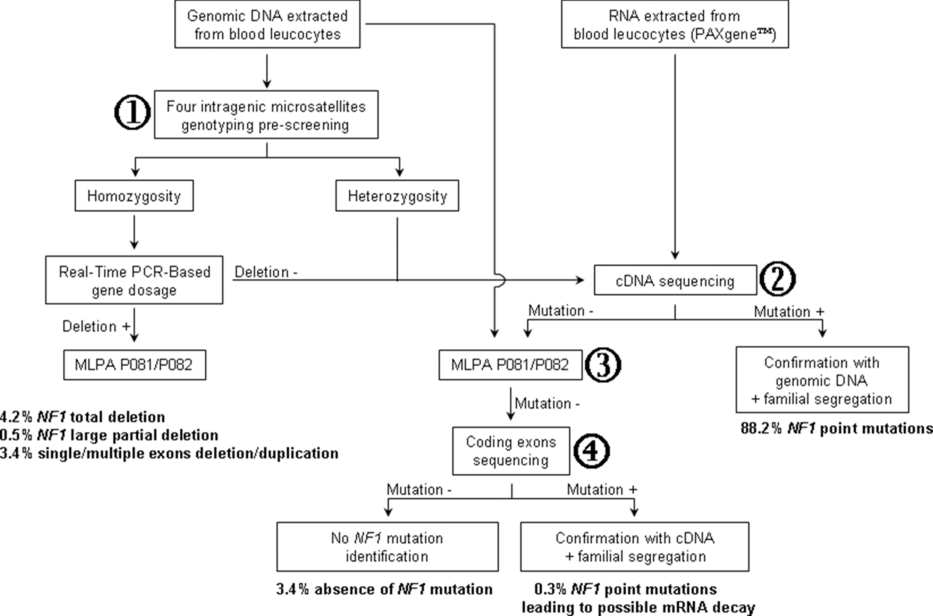
Most neurofibromatosis type 1 patients have private loss-of-function mutations scattered along the NF1 gene. Here, we present an original combined cDNA/DNA NF1 investigation strategy and report a comprehensive mutation analysis of 565 unrelated patients from the NF-France Network. A NF1 mutation was identified in 546 of the 565 patients, giving a mutation detection rate of 97%.
Coffin–Siris Syndrome and the BAF Complex: Genotype–Phenotype Study in 63 Patients
- Pages: 1519-1528
- First Published: 08 August 2013
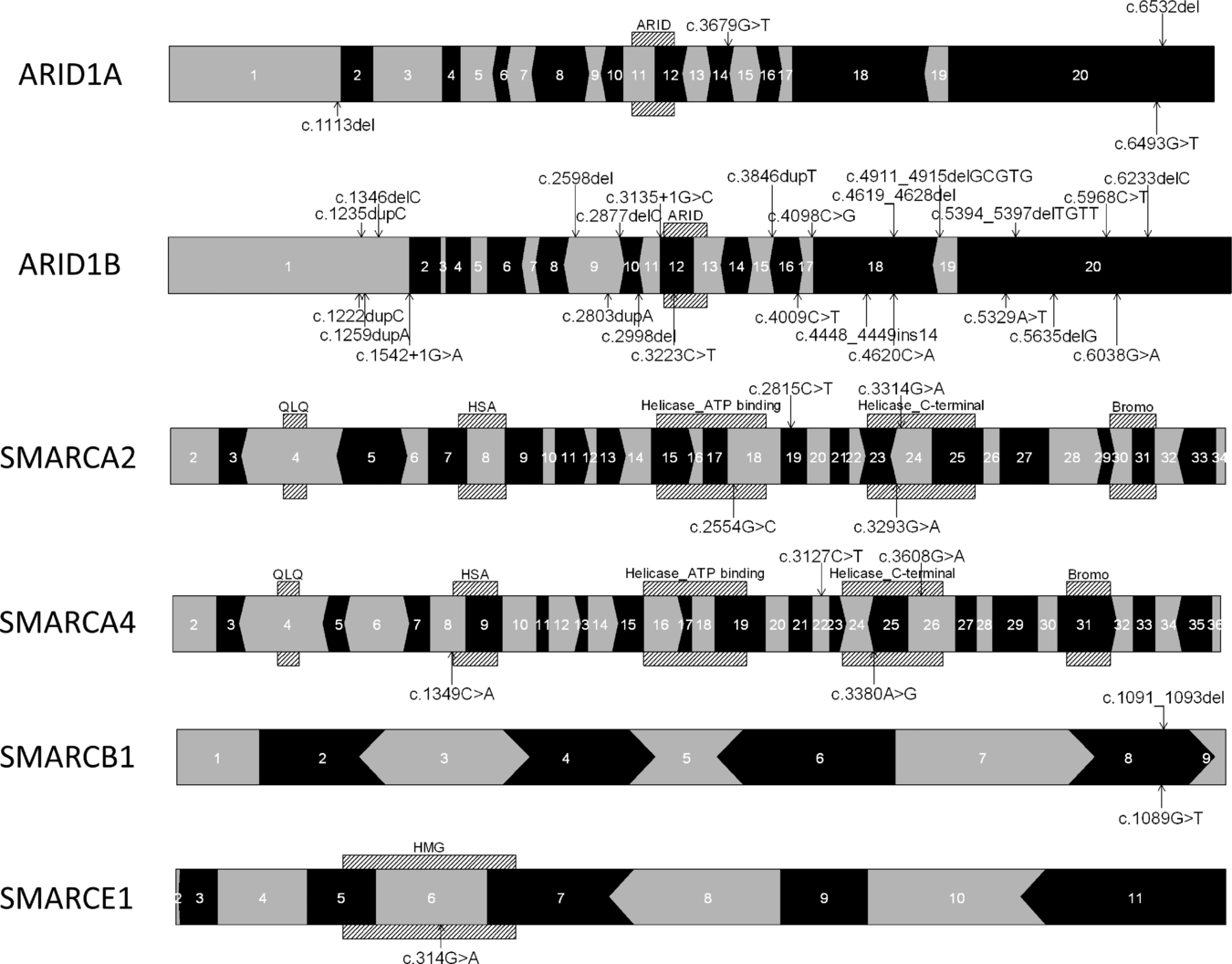
This paper describes 63 patients diagnosed with Coffin-Siris syndrome, identifying a pathogenic variant in 45. We present a genotype-phenotype correlation and provide some recommendations for the clinical care of CSS patients. We show using EVS data that especially for SMARCA2, SMARCA4 and SMARCE1 it is difficult to discriminate between non-pathogenic and pathogenic variants without access to parental DNA, and we recommend analysing parental samples.
Ferroportin Diseases: Functional Studies, a Link Between Genetic and Clinical Phenotype
- Pages: 1529-1536
- First Published: 13 August 2013
We investigated the cellular effects of five ferroportin gene (SLC40A1, NM-014585.5) mutations found in iron overloaded patients, having a potential deleterious impact from in silico analysis. We compared functional study results with patients' bioclinical phenotypes and showed that functional studies are useful tools to decipher the real impact of ferroportin mutations.
Screening of a Large Cohort of Leber Congenital Amaurosis and Retinitis Pigmentosa Patients Identifies Novel LCA5 Mutations and New Genotype–Phenotype Correlations
- Pages: 1537-1546
- First Published: 14 August 2013
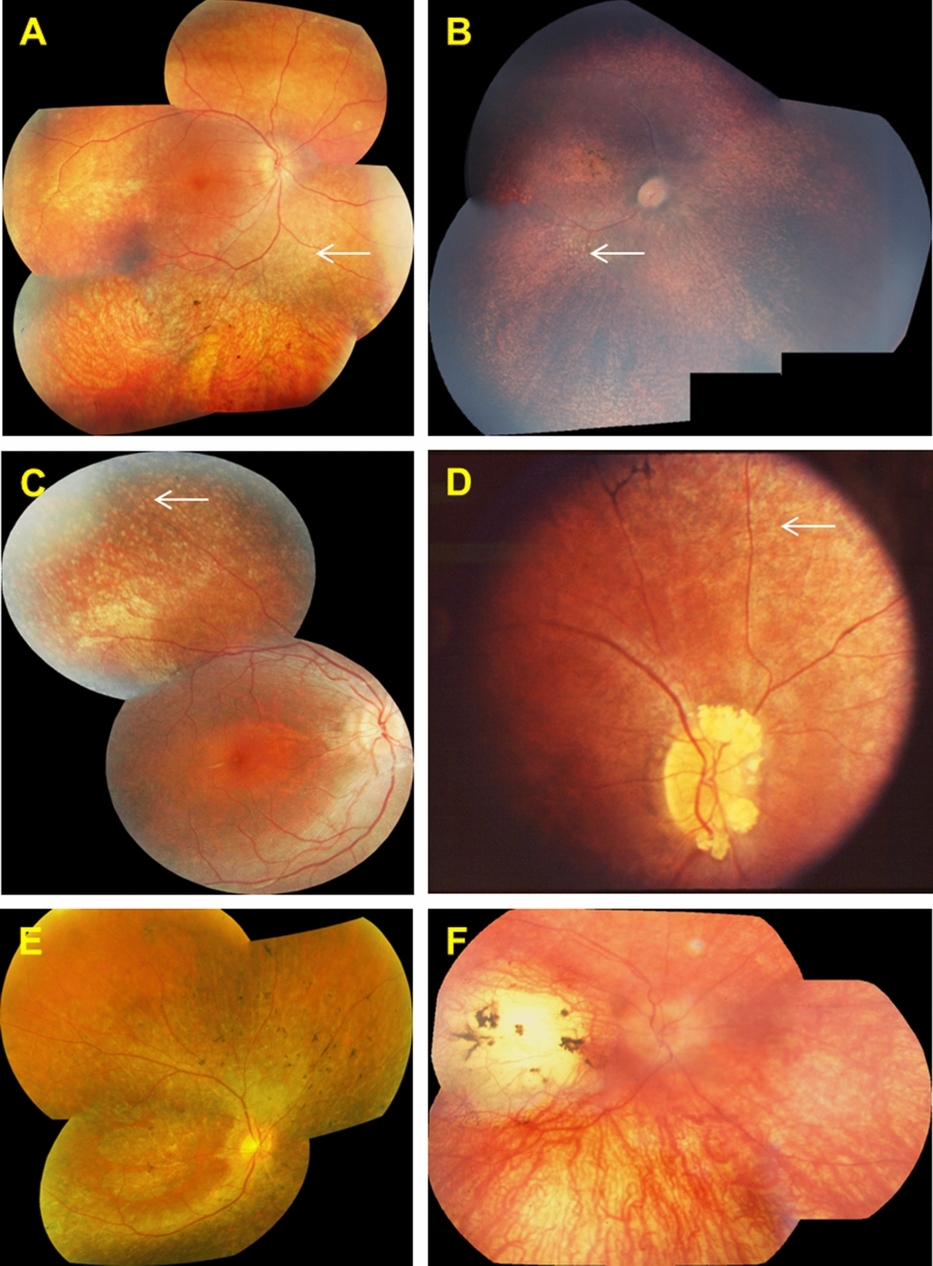
With an international LCA5 consortium, our goal was to identify LCA and RP patients with LCA5 mutations in preparation for a phase 1 clinical trial to test LCA5 gene replacement. This effort was successful in identifying 18 new LCA patients, novel mutations, and intact foveal cones, boding well for our trial.
Functional Analysis of a Large set of BRCA2 exon 7 Variants Highlights the Predictive Value of Hexamer Scores in Detecting Alterations of Exonic Splicing Regulatory Elements
- Pages: 1547-1557
- First Published: 26 August 2013
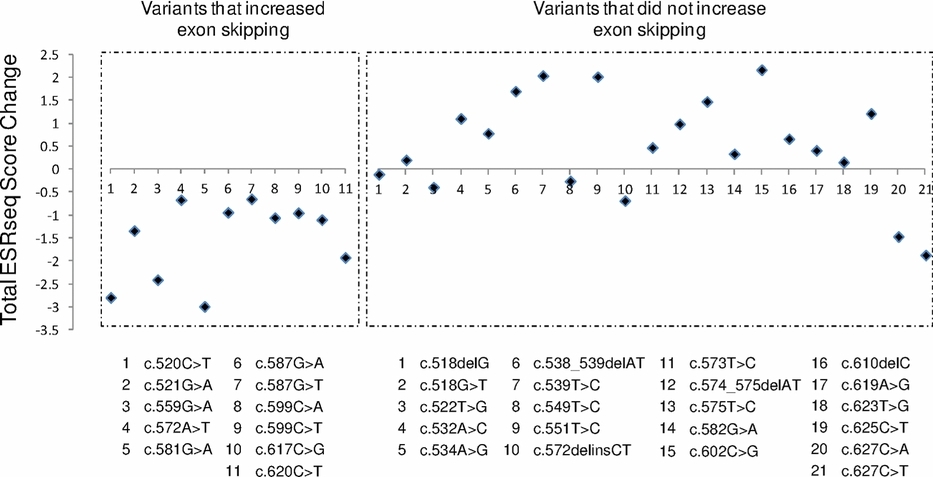
Exonic variants can alter pre-mRNA splicing not only by directly changing splice site sequences but also by modifying splicing regulatory elements, effects that are often difficult to predict. This work, based on minigene assays, revealed that 11 out of the 36 sequence variants identified in BRCA2 exon 7 are splicing regulatory mutations. We also found that the impact of these mutations could be efficiently predicted by a new in silico approach based on recently established hexamer scores.
Position of Glycine Substitutions in the Triple Helix of COL6A1, COL6A2, and COL6A3 is Correlated with Severity and Mode of Inheritance in Collagen VI Myopathies
- Pages: 1558-1567
- First Published: 29 August 2013
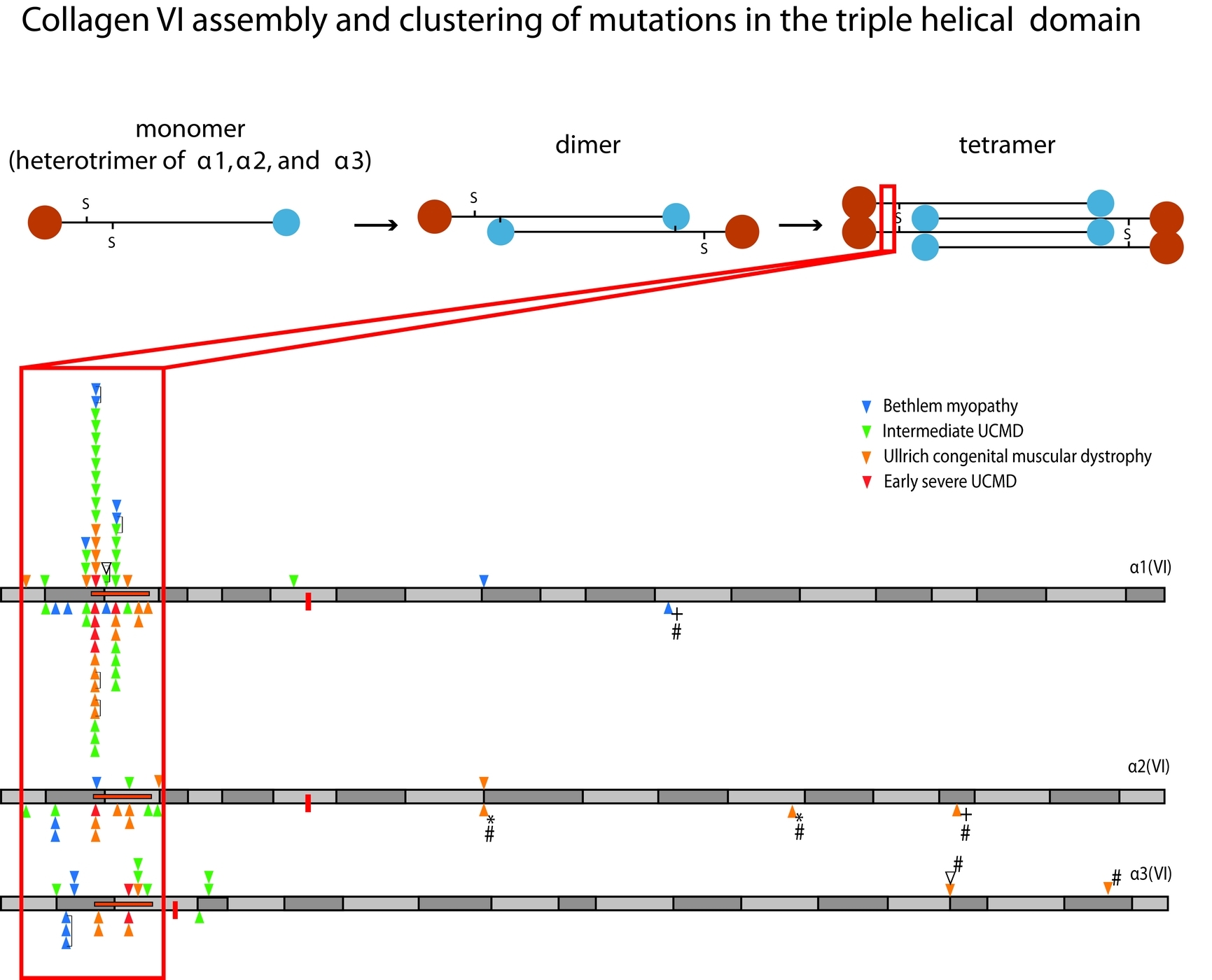
Glycine substitutions in the Gly-X-Y motif in the triple helical domain of COL6A1, COL6A2, or COL6A3 are the most commonly identified mutations in Ullrich congenital muscular dystrophy and Bethlem myopathy. We describe clinical and genetic characteristics of 97 individuals with glycine substitutions in this region including clustering of these mutations in a short segment N-terminal to the 17th Gly-X-Y triplet which is associated with increased severity. We hypothesize that this region may represent a functional domain within the triple helix.
Sensitive Detection of KRAS Mutations Using Enhanced-ice-COLD-PCR Mutation Enrichment and Direct Sequence Identification
- Pages: 1568-1580
- First Published: 23 August 2013
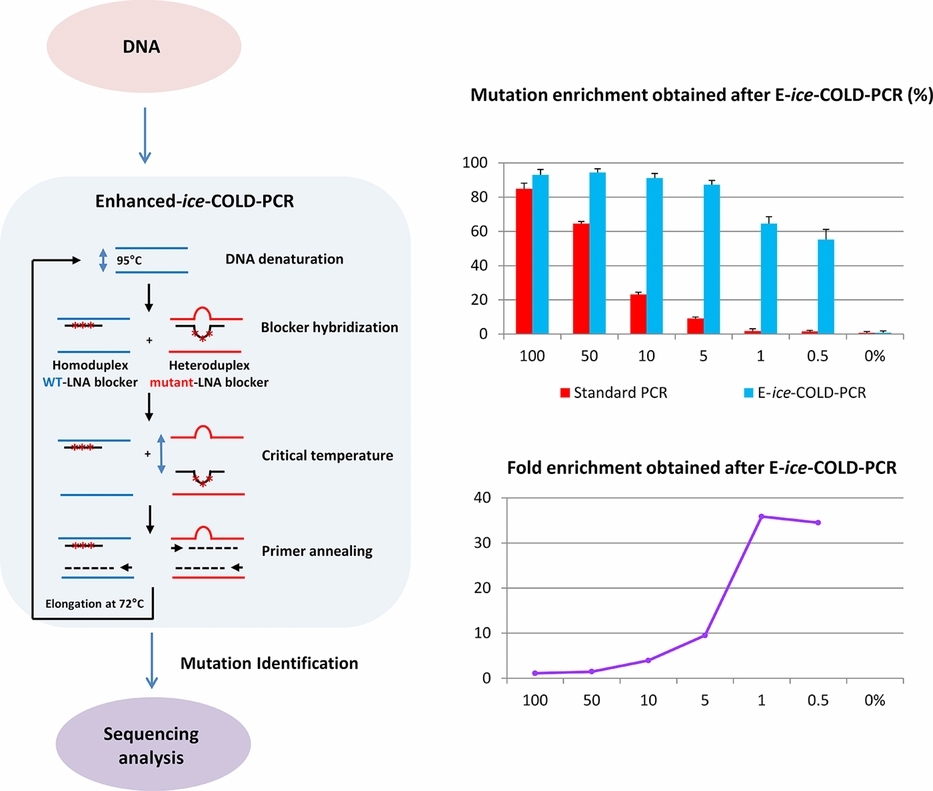
Enhanced-ice-COLD-PCR allows the enrichment, detection, and sequence-based identification of the six most frequent KRAS mutations. The method is based on a non-extendable LNA blocker sequence, complementary to the wild type sequence leading to the enrichment of mutated sequences and permitting the reliable detection of 0.1 % mutated sequences. A single genotyping assay directly following Enhanced-ice-COLD-PCR identifies the mutated base. This developed method is rapid and cost-effective and enables the detection and sequence identification of KRAS mutations within three hours.
Best Practices for Evaluating Mutation Prediction Methods
- Pages: 1581-1582
- First Published: 17 August 2013
Clinical Applications of Next-Generation Sequencing: The 2013 Human Genome Variation Society Scientific Meeting
- Pages: 1583-1587
- First Published: 17 August 2013