
Molecular Genetics & Genomic Medicine

Journal list menu

 Issue
IssueMolecular Genetics & Genomic Medicine: Volume 8, Issue 9
September 2020
Export Citations
Download PDFs
FEATURED COVER
Cover
- First Published: 14 September 2020
The cover image is based on the Original Article Genetic determinants of circulating galectin-3 levels in patients with coronary artery disease by Semon Wu et al., https://doi.org/10.1002/mgg3.1370
Cover
- First Published: 14 September 2020

The cover image is based on the Original Article Interpretation challenges of novel dual-class missense and splice-impacting variant in POLR3A-related late-onset hereditary spastic ataxia by Joel A. Morales-Rosado et al., https://doi.org/10.1002/mgg3.1341
ISSUE INFORMATION
ORIGINAL ARTICLES
Global research on Fabry's disease: Demands for a rare disease
- First Published: 07 February 2020
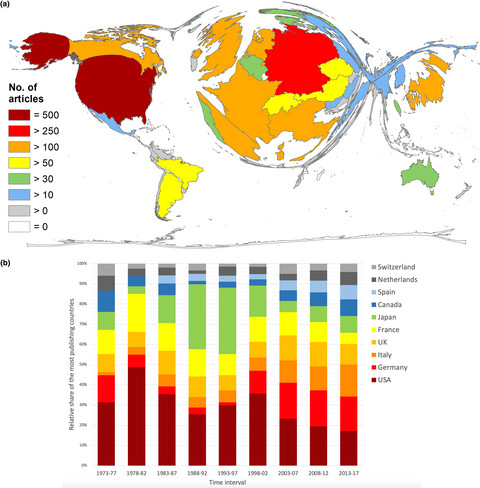
In general, high-income countries publish comparably more on Fabry disease (FD) than low- or middle-income economies. The countries' financial- and infrastructural background as well as the presence of pharmaceutical companies are unveiled as crucial factors for the FD research activity. Therefore, our findings stress that collaborative efforts should be strengthened worldwide to facilitate a better understanding of FD in terms of epidemiological background, diagnosis, and treatment and to also develop standardized guidelines to improve patient outcomes.
A homozygous missense variant of SUMF1 in the Bedouin population extends the clinical spectrum in ultrarare neonatal multiple sulfatase deficiency
- First Published: 12 February 2020
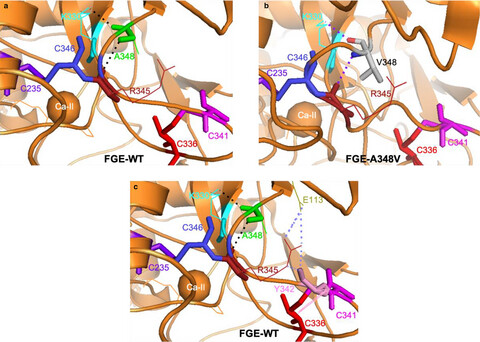
Multiple sulfatase deficiency (MSD) is an ultra-rare congenital disorder caused by SUMF1 dysfunction and often misdiagnosed due to its complex clinical presentation. Here we report two Israeli patients with a neonatal MSD caused by a likely-founder mutation SUMF1-A348V, in the Bedouin population. We show that A348V mutation leads to loss of stability of SUMF1 resulting in loss of function. The presented clinical and biochemical findings expand the spectrum of MSD, adding data on the rare neonatal severe type of MSD.
Identification of two compound heterozygous VPS13A large deletions in chorea-acanthocytosis only by protein and quantitative DNA analysis
- First Published: 14 February 2020

Chorea-acanthocytosis (ChAc; OMIM #200150) is a rare autosomal recessive condition that is caused by mutations in VPS13A. We identified two compound heterozygous VPS13A large deletions in chorea-acanthocytosis only by protein and quantitative DNA analysis. Genomic DNA (gDNA) sequencing approaches did not identify neither deletions and instead yielded false negative results.
Problems in variation interpretation guidelines and in their implementation in computational tools
- First Published: 11 March 2020
Problems and remedies are indicated for widely used variation interpretation guidelines from ACMG/AMP and AMP/ASCO/CAP and ESHG.
The phenotype-driven computational analysis yields clinical diagnosis for patients with atypical manifestations of known intellectual disability syndromes
- First Published: 26 April 2020
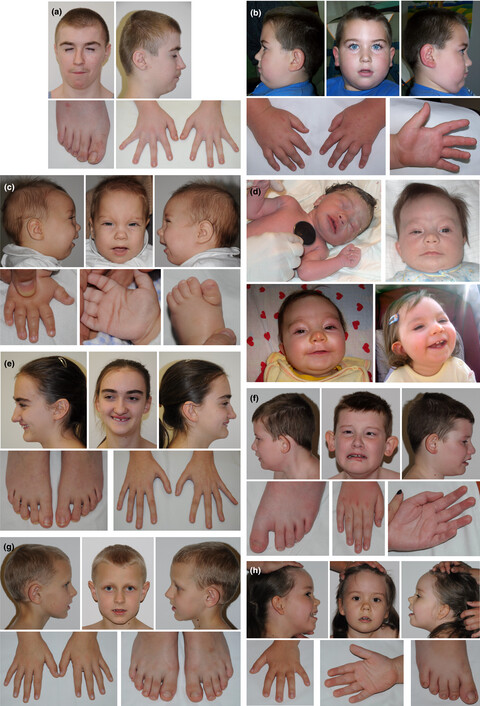
Due to extensive clinical and genetic heterogeneity of intellectual disability syndromes (ID), the process of diagnosis is very challenging even for expert clinicians. Despite recent advancements in molecular diagnostics methodologies, significant fraction of ID patients remains without clinical diagnosis. Here, in a prospective study on a cohort of 21 families (trios) with a child presenting with ID of unknown etiology, we executed phenotype-driven bioinformatic analysis method, PhenIX, utilizing targeted next generation sequencing (NGS) data and Human Phenotype Ontology (HPO)-encoded phenotype data. This approach resulted in clinical diagnosis for eight individuals presenting with atypical manifestations of syndromes with intellectual disability.
Reclassification of genetic variants in children with long QT syndrome
- First Published: 08 May 2020
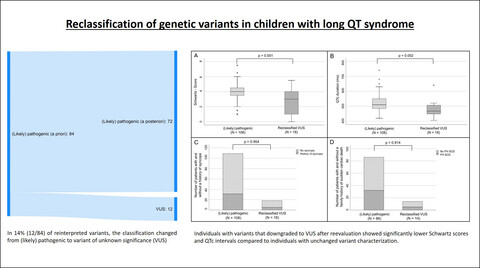
We aimed to evaluate the rate of change in variant classification based on current interpretation standards and dependent on clinical findings in children with LQTS. In 12 out of 84 reinterpreted variants (14.3%), classification changed from (likely) pathogenic to variant of unknown significance (VUS). Individuals with variants that downgraded to VUS after reevaluation showed significantly lower Schwartz scores and QTc intervals compared to individuals with unchanged variant characterization.
Functional polymorphisms of the mineralocorticoid receptor gene NR3C2 are associated with diminished memory decline: Results from a longitudinal general-population study
- First Published: 18 June 2020
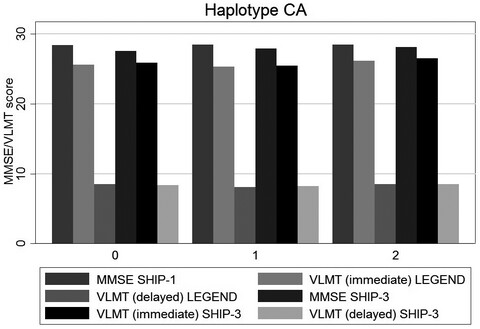
The mineralocorticoidreceptor has a central role in the brains stress response. In this longitudinal general population study, we investigated whether rs5522 and rs2070951 of the MR gene are associated with memory performance. Using two different measures for memory performance, we found that these single nucleotide polymorphisms were associated with diminished declarative memory decline.
CHD7 missense variants and clinical characteristics of Chinese males with infertility
- First Published: 22 June 2020
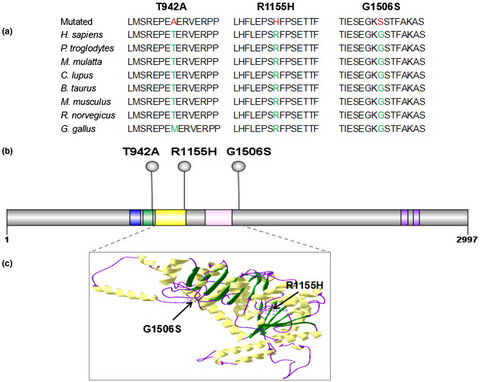
Chromodomain helicase DNA-binding protein 7 (CHD7) is one of the reported more commonly genes related to KS and IHH. We speculated that CHD7 variants may have an association with male infertility. These variants are easier to find in patients with azoospermia and severe oligospermia whose testosterone levels are decreased.
Expanded carrier screening in Chinese patients seeking the help of assisted reproductive technology
- First Published: 23 June 2020
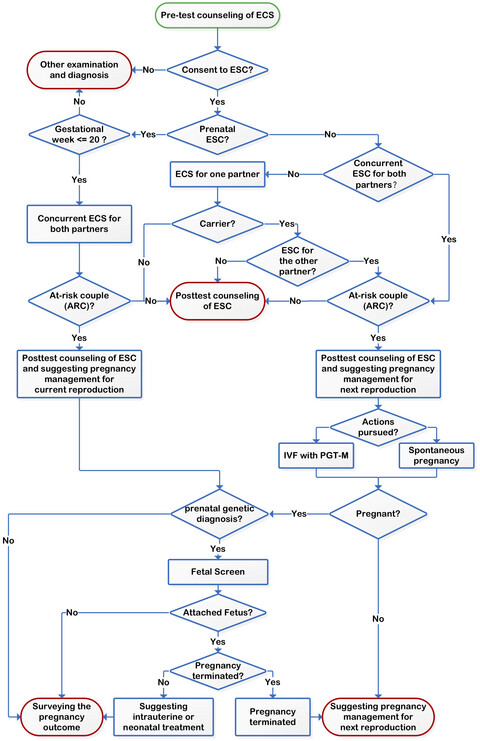
We launched a pilot population based expanded carrier screening(ECS) for 135 severe recessive Mendelian conditions in Chinese patients seeking the help of assisted reproductive technology (ART). Our study demonstrates that ESC yields a significant clinical value for the ART patients in China. In addition, by estimating the yields of the ECS panel, we identify genes that are appropriate for screening the Han population.
Cytogenetic and FISH analysis of 93 multiple myeloma Moroccan patients
- First Published: 23 June 2020
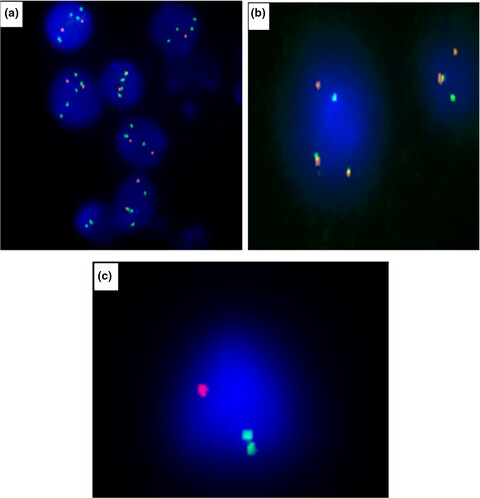
It's the first study of its kind, which studies the cytogenetic profile of multiple myeloma on Moroccan patients. In order to determine the frequency of the most recurrent anomalies in our population and their risk stratification based on these features.
Genetic determinants of circulating galectin-3 levels in patients with coronary artery disease
- First Published: 23 June 2020
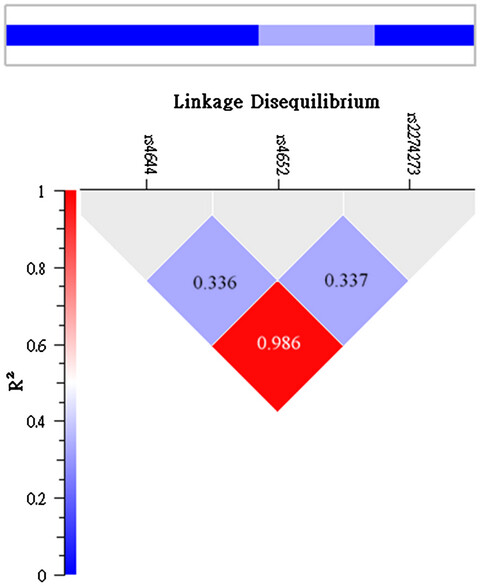
Galectin-3 levels significantly correlated with leukocyte counts, C-reactive protein, soluble intercellular adhesion molecule-1, and matrix metalloproteinase 9 levels while LGALS3 SNPs rs2274273 and rs4644 genotypes independently associated with and contributed to 20.8% of galectin-3 levels in patients with coronary artery disease. The absence of a significant association between LGALS3 gene variants and the inflammation markers levels suggested that it should be cautious when galectin-3 was used as a therapeutic target for chronic inflammatory disorders, such as coronary artery disease.
Disrupted minor intron splicing is prevalent in Mendelian disorders
- First Published: 23 June 2020
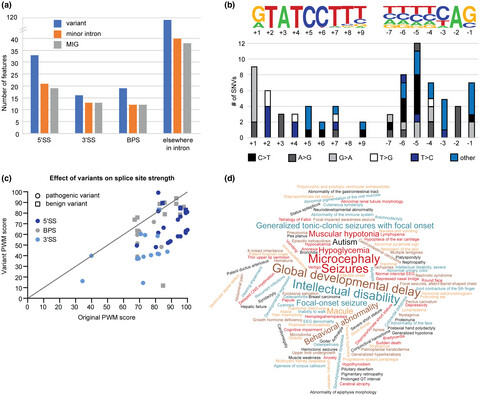
Pathogenic variants in minor spliceosome components result in disrupted splicing of minor introns and a wide range of symptoms affecting many organ systems. Here, we identified a subset of minor intron-containing genes that may play a role in the pathogenesis of these minor spliceosome-related diseases. Moreover, we report the presence of 51 pathogenic variants in minor intron splice sites that result in a reduced splice site strength.
Deleterious mis-splicing of STK11 caused by a novel single-nucleotide substitution in the 3′ polypyrimidine tract of intron five
- First Published: 23 June 2020
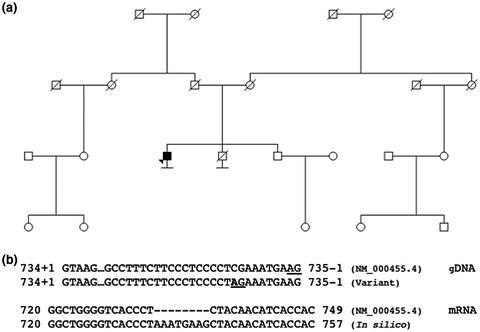
We report a novel single-nucleotide substitution c.735-10C>A in STK11 associated with Peutz–Jeghers syndrome. RNA-Seq analysis confirmed a strong, deleterious splicing effect of the variant.
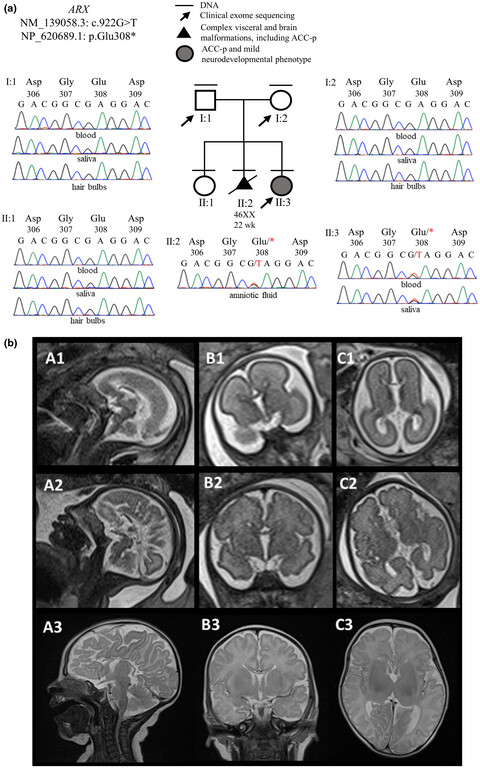
Inherited and de novo biallelic pathogenic variants in COL11A1 result in type 2 Stickler syndrome with severe hearing loss
- First Published: 24 June 2020
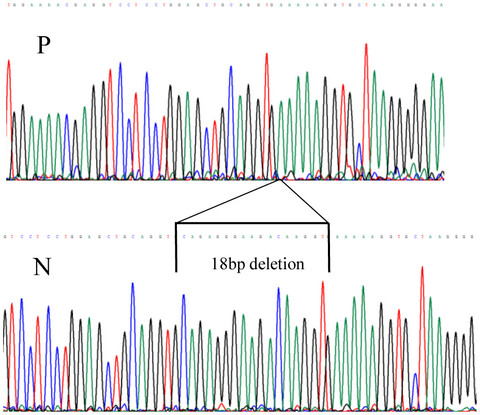
We describe an individual with a de novo in frame deletion of COL11A1 consistent with dominantly inherited Stickler syndrome but also a second inherited variant affecting normal splicing of exon 9. Allele specific RT-PCR showed that these two changes are bi-allelic. Like the previously reported recessive cases involving exon 9 variants, this patient also had a phenotype of Stickler syndrome with severe hearing loss and also more severe ocular features.
Different prevalence of T2DM risk alleles in Roma population in comparison with the majority Czech population
- First Published: 24 June 2020
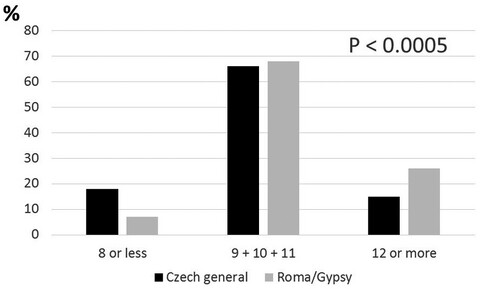
The frequency of most T2DM-associated genotypes differed significantly between the Czech majority population and Roma. Roma subjects have in mean increased numbers of risky alleles. Likely, genes are in background of the increased T2DM incidence in this ethnic group.
The study of METTL14, ALKBH5, and YTHDF2 in peripheral blood mononuclear cells from systemic lupus erythematosus
- First Published: 25 June 2020
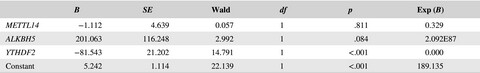
SLE patients had lower mRNA expression of METTL14, ALKBH5 and YTHDF2 and logistic regression analysis revealed that decreased mRNA expression of YTHDF2 was a risk factor for SLE
Determination of mutations in iron regulating genes of beta thalassemia major patients of Khyber Pakhtunkhwa, Pakistan
- First Published: 25 June 2020
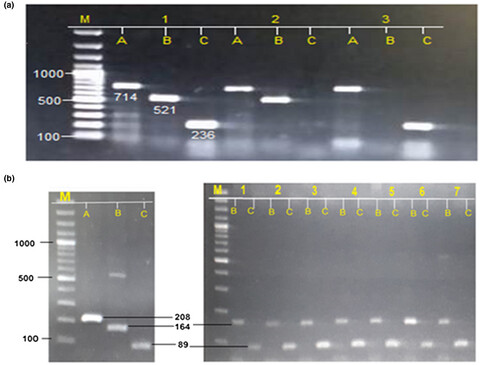
We studied the presence of G71D mutation of HAMP gene and H63D mutation of HFE gene in beta thalassemia major and minor group to check the association of these mutations with serum ferritin level of beta thalassemia patients.
A novel splicing pathogenic variant in COL1A1 causing osteogenesis imperfecta (OI) type I in a Chinese family
- First Published: 25 June 2020
We use Whole exome sequencing identify a novel splicing pathogenic variant in COL1A1 gene causing osteogenesis imperfecta (OI) type I in a Chinese family.
FARP-1 deletion is associated with lack of response to autism treatment by early start denver model in a multiplex family
- First Published: 25 June 2020
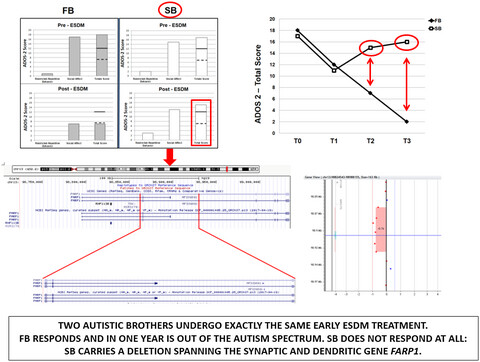
This is the first study addressing the involvement of FARP1 in a human disorder and the genetic basis of response to early behavioral intervention in child psychiatry. Though limited to one family, these results are a paradigmatic example of how genetic variants may represent useful treatment markers, able to aid clinicians in predicting response and in prescribing the therapeutic approach most likely to succeed in the single patient.
Rare variants in the GABAA receptor subunit ε identified in patients with a wide spectrum of epileptic phenotypes
- First Published: 25 June 2020
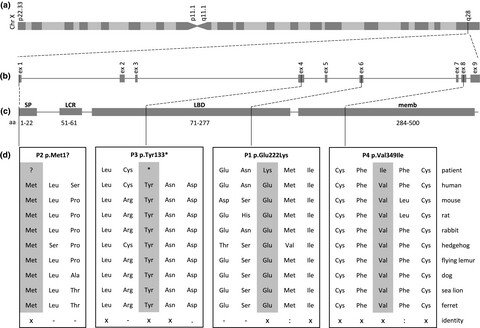
The neural amino acid y-aminobutyric acid (GABA) regulates activity of channel pores by binding to transmembrane GABA-receptors (GABRs). We identified four hemizygous sequence variants in the GABAA receptor subunit ε gene (GABRE) in four epileptic patients. Our clinical and molecular genetic findings suggest that GABRE is a likely candidate gene for epilepsy.
Systematic summarization of the expression profiles and prognostic roles of the dishevelled gene family in hepatocellular carcinoma
- First Published: 26 June 2020
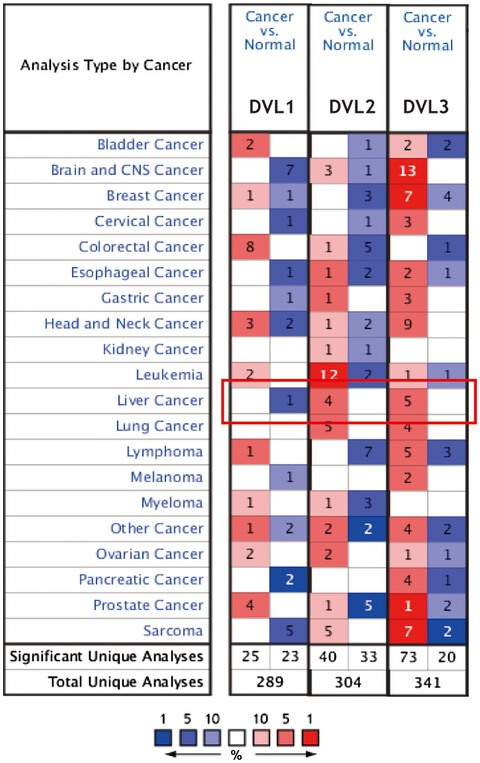
Our study systematically summarized the expression profiles and prognostic values of the DVL family members in HCC. The high expression of DVLs mRNA was significantly related to the poor prognosis of patients with HCC. Besides, the results also revealed DVLs expression level was correlated with the infiltration levels of multiple immune cells. Therefore, detection of the expression levels of DVLs in HCC tissues might be used as a novel strategy to predict the prognosis of patients with HCC.
Investigation on the role of biallelic variants in VEGF-C found in a patient affected by Milroy-like lymphedema
- First Published: 26 June 2020
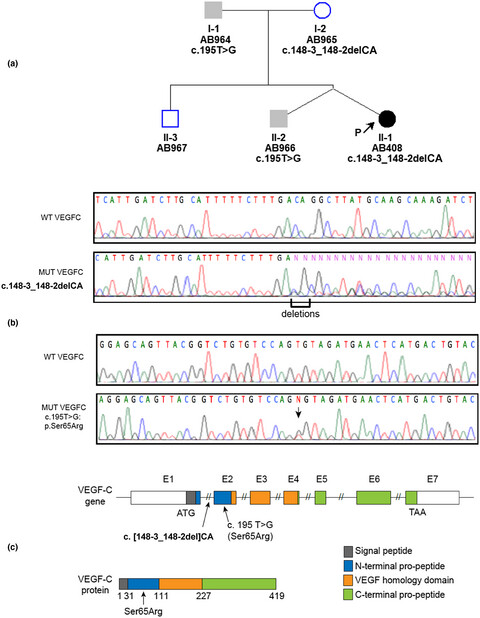
A genetic and biochemical analysis of VEGF-C variations were performed on a female proband affected by primary lymphedema of the right lower limb and on her entire family. Biallelic variants of VEGF-C variations were found in the proband: a novel p.(Ser65Arg) and a pathogenic c.148-3_148-2del, of paternal and maternal origin, respectively. Clinical examination of the family by lymphoscintigraphy as well as biochemical analysis show that both variants are required to the development of the proband Milroy-like lymphedema.
TP53 p.Arg337His geographic distribution correlates with adrenocortical tumor occurrence
- First Published: 27 June 2020
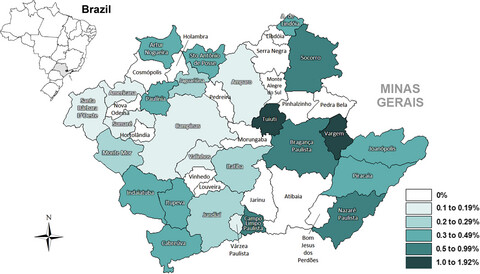
The study presents a reliable and efficient method for high-throughput TP53 p.Arg337His genotyping, what allowed authors to determine the frequency and geographic distribution of this mutation in a cohort from the most populous state in Brazil. In addition, adrenocortical tumor was found to be a good sentinel cancer to suppose p.Arg337His presence in the region analyzed.
Interpretation challenges of novel dual-class missense and splice-impacting variant in POLR3A-related late-onset hereditary spastic ataxia
- First Published: 29 June 2020

The article highlights a POLR3A-related adult-onset spastic ataxia case and describe a novel dual-class missense and splicing variant. Follow-up RNA sequencing revealed a complex disruption leading to four RNA transcript products, each with different functional impacts.
SHORT syndrome in two Chinese girls: A case report and review of the literature
- First Published: 29 June 2020
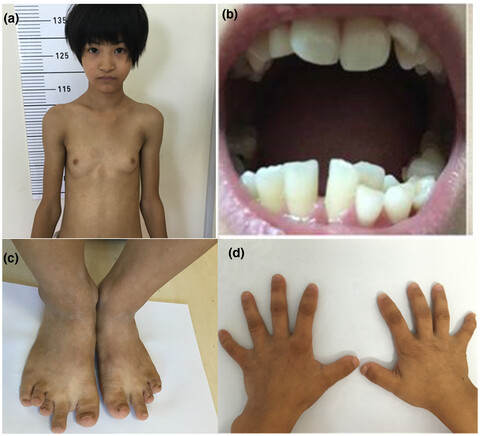
SHORT syndrome is a rare inherited multisystem disease that includes characteristic facial features, growth retardation and metabolic anomalies and is related to heterozygous mutations in the PIK3R1 gene. To date, ten different mutations in the PIK3R1 gene have been reported. After searching the literature, we found that there were correlations between genotype in PIK3R1 and the phenotype of SHORT syndrome to a certain extent.
A Korean child diagnosed with malonic aciduria harboring a novel start codon mutation following presentation with dilated cardiomyopathy
- First Published: 30 June 2020

This is the first report of MA diagnosed in a patient following presentation with infantile-onset dilated cardiomyopathy following negative malonylcarnitine measurement results in a newborn screening program. The disease was successfully managed with a combination of medical therapy and dietary intervention.
Overexpression of SNTG2, TRAF3IP2, and ITGA6 transcripts is associated with osteoporotic vertebral fracture in elderly women from community
- First Published: 30 June 2020
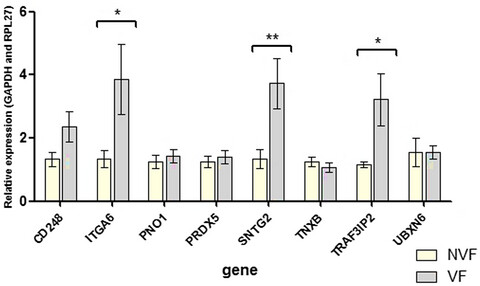
Our data identified and validated the association of SNTG2, TRAF3IP2, and ITGA6 with osteoporotic vertebral fracture (VF) in elderly women, independently of bone mineral density. These results suggest that these transcripts have potential clinical significance and may help to explain the molecular mechanisms and biological functions of VF.
Identification and functional characterization of a novel surfactant protein A2 mutation (p.N207Y) in a Chinese family with idiopathic pulmonary fibrosis
- First Published: 30 June 2020

Mutation in SFTPA2 was rare reported, and we identified a novel pathogenic mutation (p.N207Y) of SFTPA2 gene in idiopathic pulmonary fibrosis (IPF) patients in this study. Functional study further confirmed the novel mutation may affect the expression and secretion of surfactant protein A2 protein and induce endoplasmic reticulum stress and apoptosis. Our study not only confirmed the importance of SFTPA2 in IPF, but also expanded the spectrum of SFTPA2 mutations and contributed to the genetic diagnosis and counseling of IPF patients.
Determining the best candidates for next-generation sequencing-based gene panel for evaluation of early-onset epilepsy
- First Published: 01 July 2020
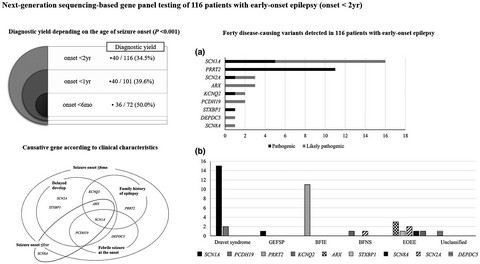
Next-generation sequencing-based targeted gene panel is an effective diagnostic test for early-onset epilepsy with heterogeneous genotypes and phenotypes. The best candidates for genetic testing are patients with earlier seizure onset and family history of epilepsy.
The expression and regulation of HOX genes and membrane proteins among different cytogenetic groups of acute myeloid leukemia
- First Published: 02 July 2020
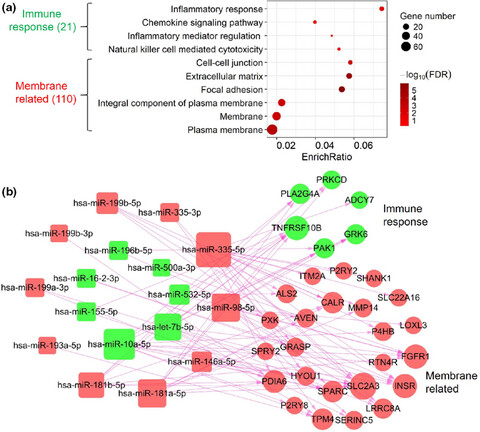
We integrated comprehensive bioinformatics analyses for genes between favorable, poor and cytogenetically normal (CN) groups of acute myeloid leukemia (AML) patients. We found that differentially expressed genes were enriched in membrane related processes. Eleven genes and two microRNAs were significantly differentially expressed among these 3 AML groups. Six HOXA and three HOXB genes was significantly in low expression and high methylation in AML with favorable cytogenetics.
Aberrant X chromosomal rearrangement through multi-step template switching during sister chromatid formation in a patient with severe hemophilia A
- First Published: 05 July 2020
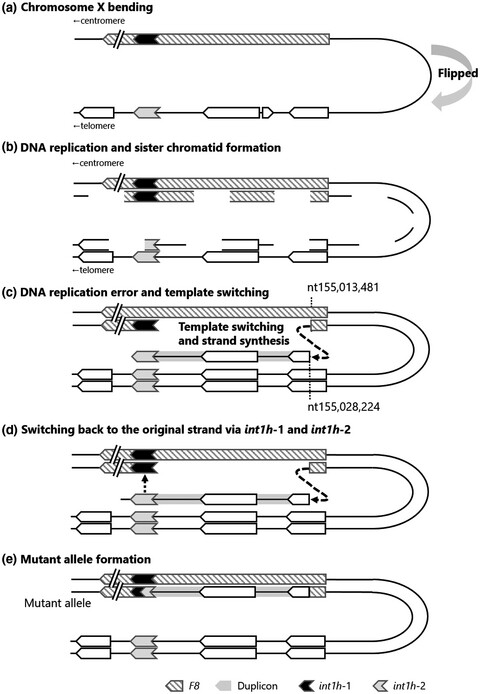
Hemophilia A (HA) is an X-linked recessive bleeding disorder caused by mutations in the coagulation factor VIII gene (F8). We identified the aberrant chromosome X with a split F8 due to a multi-step rearrangement in a severe HA patient. The rearrangement mechanism was suggested as a multi-step rearrangement with template switching such as FoSTeS/MMBIR and/or homologous sequence associating recombination during a sister chromatid formation.
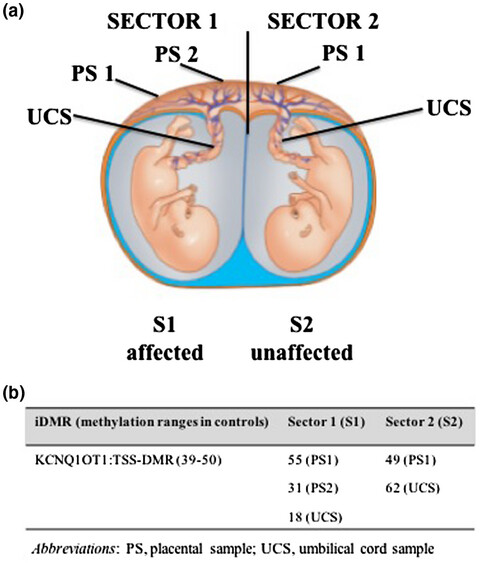
(Epi)genetic profiling of extraembryonic and postnatal tissues from female monozygotic twins discordant for Beckwith–Wiedemann syndrome
- First Published: 06 July 2020
Spectrum of gene mutations identified by targeted next-generation sequencing in Chinese leukemia patients
- First Published: 07 July 2020
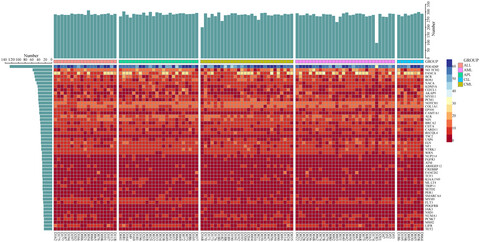
We identified a total of 4,655 single-nucleotide variants and 614 insertions and deletions in 419 genes, in which PDE4DIP, NOTCH2, FANCA, BCR and ROS1 emerged as the highly mutated genes. Based on American College of Medical Genetics and Genomics guidelines, 39 pathogenic and likely pathogenic mutations in 27 genes were found. Our study provided a map of gene mutations in Chinese patients with leukemia and gave insights into the molecular pathogenesis of leukemia.
Clinical features and genetic characteristics of homozygous spinocerebellar ataxia type 3
- First Published: 09 July 2020
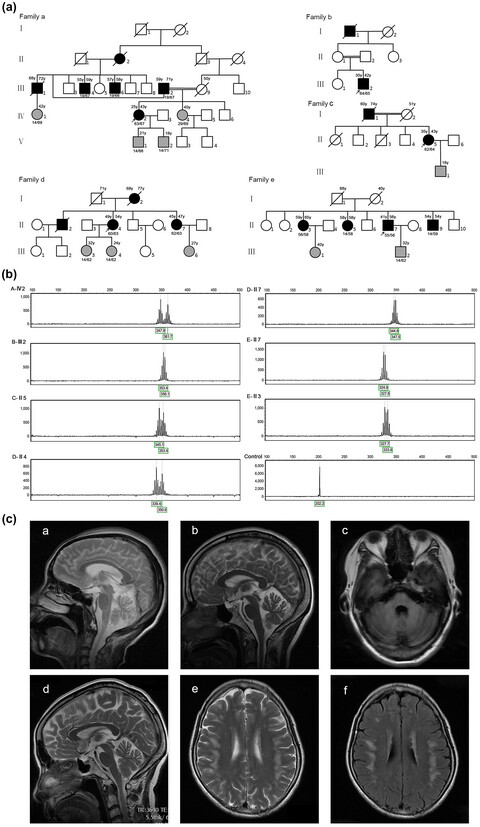
We analyzed seven newly homozygous spinocerebellar ataxia type 3 patients from five families and 14 homozygotes reported previously. The AAO was significantly inversely correlated with both the large and small expanded CAG repeats (r = −.7682, p < .0001). Gene dosage effect may play an important role in the AAO and severity of disease, and homozygosity for ATXN3 enhances phenotypic severity.
Paternal gender specificity and mild phenotypes in Charcot–Marie–Tooth type 1A patients with de novo 17p12 rearrangements
- First Published: 09 July 2020
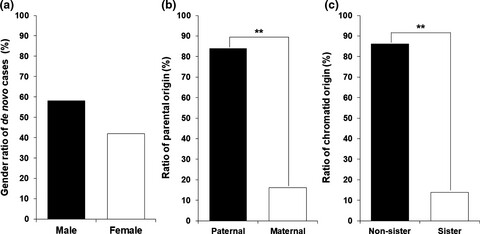
This study identified 40 de novo cases in 211 CMT1A or HNPP trio families. Paternal ages at the affected child births were slightly higher in the de novo CMT1A group than in the non-de novo CMT1A control group. For the disability score of CMTNS, the de novo CMT1A group had a slightly lower value compared to the control group. This will be the first study to suggest that de novo CMT1A patients tend to have milder symptoms and that the paternal ages at child births in the de novo group are higher than those of the non-de novo group.
Identification of paternal germline mosaicism by MicroSeq and targeted next-generation sequencing
- First Published: 09 July 2020

we reported for the first time the application of MicroSeq in the diagnosis of Mendelian hereditary disease, which was a technique combined chromosome microdissection and next-generation sequencing (NGS). We used targeted next-generation sequencing (TNGS) to assess the mosaic ratios of known site and found that 500X reads depths coverage could meet the detection requirements of low-frequency mosaic mutation of 2% to 3%.
Two novel mutations in the MCM8 gene shared by two Chinese siblings with primary ovarian insufficiency and short stature
- First Published: 11 July 2020
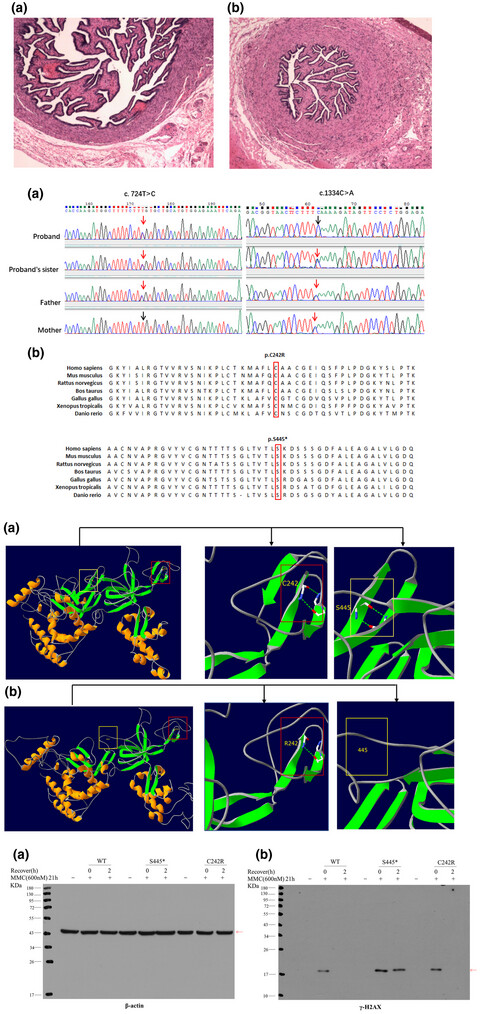
MCM8 is cause of primary ovarian insufficiency (POI) but is seldom diagnosed in adolescents and children. Affected children with novel pathogenetic mutations p.C242R and p.S445X in the MCM8 gene are characterized by POI, short stature, cancer susceptibility, and genomic instability.
Genetic profile of non-small cell lung cancer (NSCLC): A hospital-based survey in Jinhua
- First Published: 12 July 2020
We described clinical characteristics and genetic profiles of 256 patients with non-small cell lung cancer. We found that the median variant allele frequency (VAF) in adenocarcinoma was significantly lower than those in squamous cell carcinoma and the median VAF was significantly higher in stage II-IV than in stage I. Besides, we found that ALK and BRAF mutations had a strong correlation with younger age.
Parental genetic knowledge and attitudes toward childhood genetic testing for inherited eye diseases
- First Published: 13 July 2020

This study illustrated that the effective strategies should be taken to provide the correct knowledge of genetics and genetic testing to parents, especially those who need to make an informed decision thereon to undertake childhood genetic testing.
Association of genetic variants at CETP, AGER, and CYP4F2 locus with the risk of atrophic age-related macular degeneration
- First Published: 14 July 2020
We aimed to determine the frequency of the genotypes of CETP(rs5882; rs708272; rs3764261; rs1800775; rs2303790), RAGE (rs1800624; rs1800625) and CYP4F2 (rs1558139) in patients (pts) with atrophic age-related macular degeneration (AMD). We identified 2 polymorphisms with higher risk of atrophic AMD (rs5882 and rs1800625) and one protective polymorphism (rs3764261).
Identification of histone malonylation in the human fetal brain and implications for diabetes-induced neural tube defects
- First Published: 15 July 2020
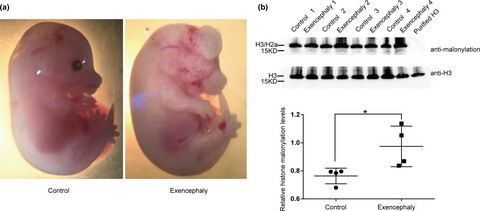
In this manuscript, we reported, for the first time, the presence of histone malonylation in human fetal brain and mouse neural stem cells.
The results reveal that histone malonylation aberrant in diabetes-induced NTDs.
These results may bring a new insight in the pathological role of histone modifications in human NTDs.
Development of an immune-related prognostic model for pediatric acute lymphoblastic leukemia patients
- First Published: 15 July 2020
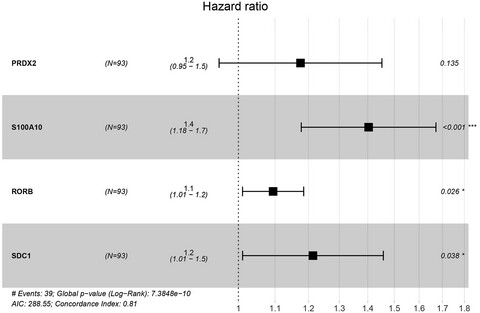
Differentially expressed immune-related genes between the relapse and non-relapse groups of 251 pediatric acute lymphoblastic leukemia patients were analyzed. Four optimal genes (PRDX2, S100A10, RORB and SDC1) were screened, and then an immune-related prognostic model based on these four genes were constructed.
THIS ARTICLE HAS BEEN RETRACTED
RETRACTED: Evaluation of a six-dye multiplex composed of 27 markers for forensic analysis and databasing
- First Published: 16 July 2020
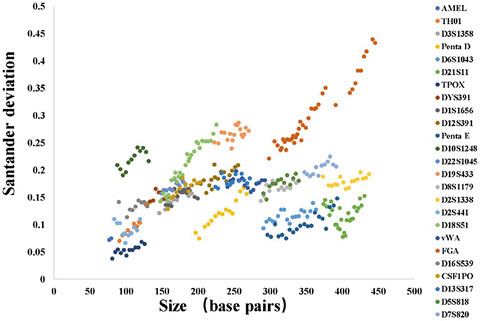
- A 6-dye multiplex composed of 27 markers were evaluated, according to SWGDAM guidelines.
- High power of discrimination and cumlative probability of paternity exculsion value in two Chinese population.
- The 6-dye multiplex is robust, reliable and suitable tool for forensic analysis and data-basing.
ORIGINAL ARTICLES
An update of pathogenic variants in ASPM, WDR62, CDK5RAP2, STIL, CENPJ, and CEP135 underlying autosomal recessive primary microcephaly in 32 consanguineous families from Pakistan
- First Published: 17 July 2020
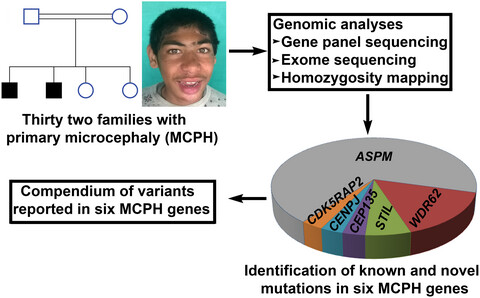
Genomic analyses of 32 Pakistani families manifesting primary microcephaly.
BRCA2 c.8827C>T pathogenic mutation in a consanguineous Chinese family with hereditary breast cancer
- First Published: 20 July 2020
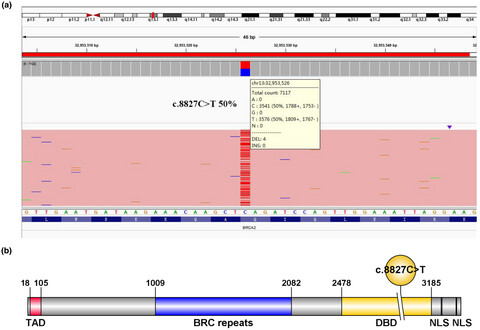
We identified a BRCA2 c.8827C>T nonsense mutation with a truncated BRCA2 protein in a consanguineous Chinese Han family through next-generation sequencing platform, suggesting individuals with this mutation should be regularly screened for malignancies such as breast, prostate, and ovarian cancer. Our study verified the function of this BRCA2 mutation site with Real-time PCR and Western blot analysis and provided a new target for the precise treatment of such patients.
CLINICAL REPORTS
From a variant of unknown significance to pathogenic: Reclassification of a large novel duplication in BRCA2 by high-throughput sequencing
- First Published: 13 November 2019
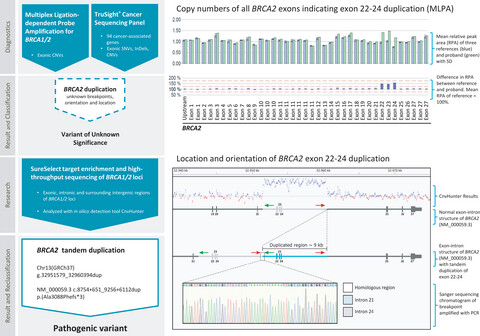
In our manuscript we report a novel BRCA2 duplication of exons 22–24 in a female patient with bilateral breast cancer. The duplicated region was initially detected by gene panel sequencing and multiplex ligation-dependent probe amplification. Since the breakpoints are located in introns, the duplication could only be further characterized by high-throughput sequencing including noncoding regions.
Medulloblastoma, macrocephaly, and a pathogenic germline PTEN variant: Cause or coincidence?
- First Published: 17 May 2020
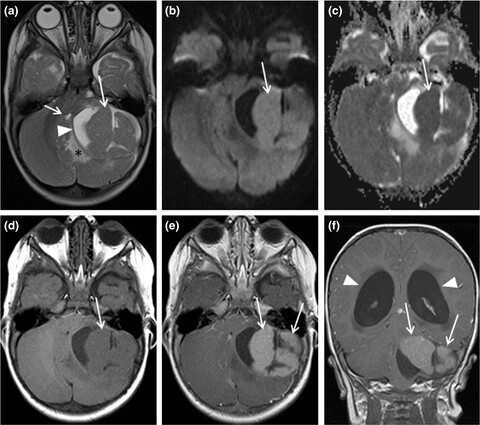
Pathogenic germline variants of the PTEN gene may predispose to medulloblastoma at an early age. We show that a patient carrying a pathogenic PTEN variant reached remission with current treatment protocols. We propose follow-up guidelines to patients shown to carry pathogenic PTEN variants.
Genetic alteration of colorectal adenoma-carcinoma sequence among gastric adenocarcinoma and dysplastic lesions in a patient with attenuated familial adenomatous polyposis
- First Published: 16 June 2020
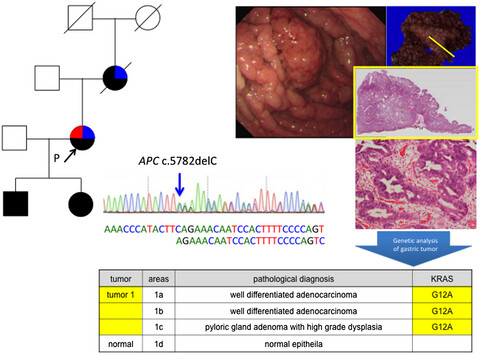
Germline APC variant c.5782delC was found by direct sequencing and somatic KRAS mutations in these tumors were identified by next-generation sequencing. The colonic adenoma-carcinoma sequence among gastric carcinoma and dysplastic lesions was indicated in Familial adenomatous polyposis-associated gastric carcinogenesis.
Variable clinical expression of Stickler Syndrome: A case report of a novel COL11A1 mutation
- First Published: 17 June 2020
A novel COL11A1 mutation identified by Whole Genome Sequencing, confirming the diagnosis of Stickler Syndrome in a child. Clinical features included early-onset of myopia with vitreal abnormalities, facial dysmorphic characteristics and mild hearing loss later in childhood.
MYH2 myopathy, a new case expands the clinical and pathological spectrum of the recessive form
- First Published: 24 June 2020

MYH2 mutations are responsible for a late-onset autosomal dominant (AD) disease characterized by reduction in type 2A muscle fibre number and size along with rimmed vacuoles, and for an early-onset recessive form characterized by complete loss of type 2A fibres. We describe a patient presenting a late-onset ophthalmoparesis and proximal limb muscle weakness, with histopathological muscle findings similar to those previously described in AD forms and a recessive MYH2 genotype. Our case report contributes to expand the clinical and genetic spectrum of MYH2 myopathies, extending the knowledge of this very rare disease.
Untypically mild phenotype of a patient suffering from Sanfilippo syndrome B with the c.638C>T/c.889C>T (p.Pro213Leu/p.Arg297Ter) mutations in the NAGLU gene
- First Published: 24 June 2020
Patient ZK, who was diagnosed for Sanfilippo syndrome B, is a compound heterozygote (c.638C>T/c.889C>T) in the NAGLU gene, with a relatively high (about 25%) residual activity of α-N-acetylglucosaminidase. She expressed relatively mild phenotype, with various cognitive and communication functions preserved, and relatively good motoric functions, at the age of 13 years. We suggest that this mild phenotype might arise from partially preserved activity of the p.Pro213Leu variant of α-N-acetylglucosaminidase, causing slow accumulation of HS and slow progress of the disease.
Monochorionic diamniotic twins of discordant external genitalia with 45,X/46,XY mosaicism
- First Published: 25 June 2020
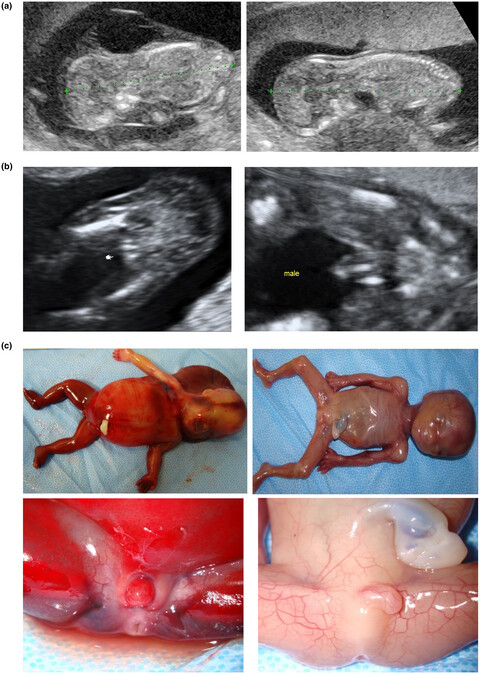
This report was case of 45,X/46,XY mosaicism in monozygotic twins with different external genitalia. Cell counting using fluorescence in situ hybridization with XY probes (XY-FISH) showed 20% and 80% abundance of 45,X cells in the internal genitalia, liver, heart, lung, adrenal gland, bone marrow, and spine of the male and female fetuses, respectively.
A 17q24.3 duplication identified in a large Chinese family with brachydactyly-anonychia
- First Published: 25 June 2020
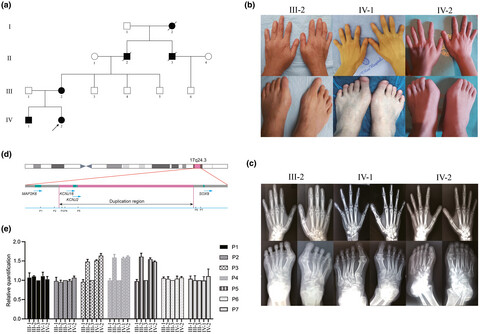
We firstly identified a new subtype of brachydactyly in a large Chinese family carrying the similar duplication involving in regulatory region of SOX9 (17q24.3), which is important for the future gene diagnosis.
Diagnostic utility of integrated analysis of exome and transcriptome: Successful diagnosis of Au-Kline syndrome in a patient with submucous cleft palate, scaphocephaly, and intellectual disabilities
- First Published: 26 June 2020
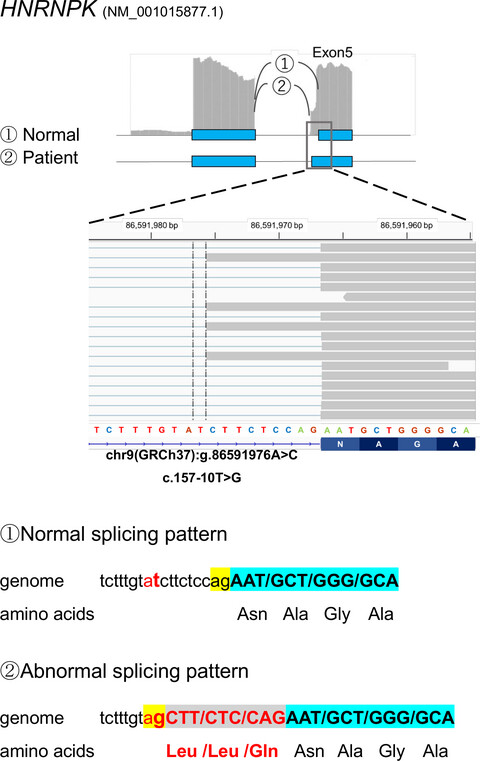
We have developed a novel analytic method, SAVNet that combines transcriptome and exome analysis which enabled the successful detection of carriers of splicing variants in the disease-causing genes of autosomal recessive disorders within a normal cohort. In this study, we documented the clinical utility of the SAVNet analysis in delineating splicing defects in patients without a diagnosis. Using this technique, we were able to detect splicing abnormalities in an undiagnosed patient and diagnose Au-Kline syndrome.
Expanding the phenotype of STRA6-related disorder to include left ventricular non-compaction
- First Published: 29 June 2020
We describe a fetus with bilateral anophthalmia, bilateral pulmonary agenesis, interrupted aortic arch type A and left ventricular non-compaction (LVNC) who was found to have a homozygous splicing variant in the STRA6 gene. This case adds to the phenotypic spectrum of STRA6-related disorder, supports the association with LVNC, and suggests that the mutation herein reported may be a hotspot in STRA6.
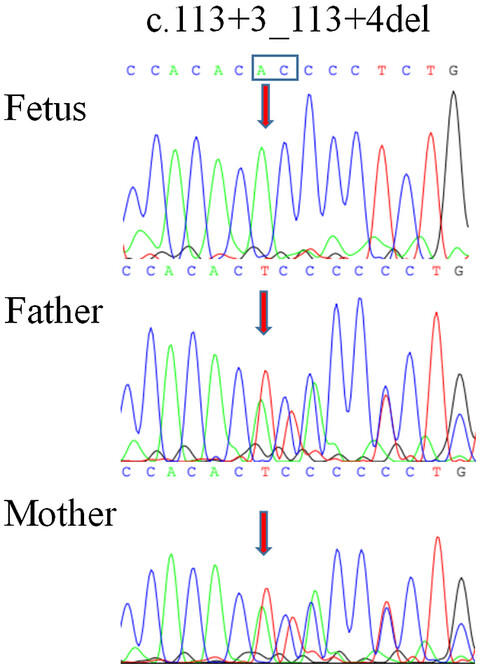
Identification of novel splice mutation in SMAD3 in two Cypriot families with nonsyndromic thoracic aortic aneurysm. Two case reports
- First Published: 29 June 2020
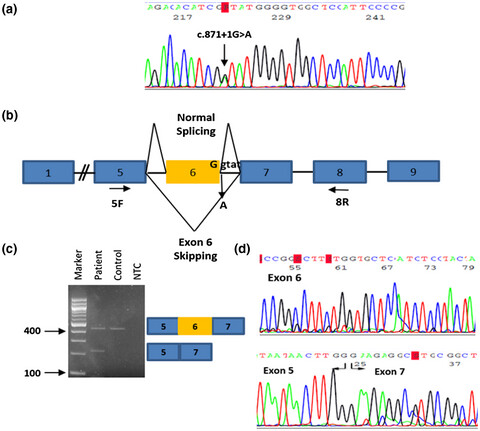
In this study we report two apparently unrelated Cypriot families with nonsyndromic familial thoracic aortic aneurysm and dissection associated with a novel variant in SMAD3. Analysis of mRNA from patients’ tissue confirmed aberrant splicing and exon 6 skipping. This study demonstrates the importance of a comprehensive clinical and genetic evaluation aimed at early diagnosis and intervention.
A novel DNAH11 variant segregating in a sibship with heterotaxy and implications for genetic counseling
- First Published: 07 July 2020
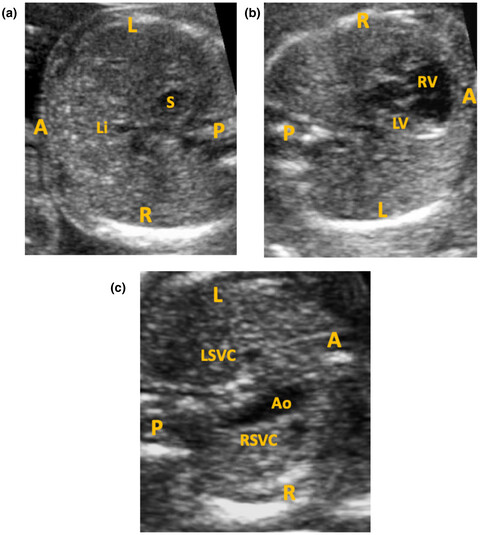
Isomerism or heterotaxy syndrome is the loss of normal asymmetry of the internal thoracoabdominal organs in the left-right axis and is associated with cardiovascular malformations. We present a novel likely pathogenic variant (c.11662C>T) in DNAH11 that has manifested in heterotaxy with variability in phenotypes for subsequent pregnancies of common parents. This report demonstrates that sibship illustrates potential variability in phenotypes associated with the same pathogenic variants within a family and highlights the difficulty in genetic counseling due to the variation in clinical presentation.
Adult-onset Krabbe disease due to a homozygous GALC mutation without abnormal signals on an MRI in a consanguineous family: A case report
- First Published: 17 July 2020
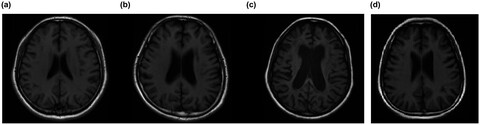
- GALC gene analysis revealed a rare homozygous c.1901T>C mutation in exon 16 of the patient.
- No abnormal signals were found in the brain MRI,but a significantly decreased GMV in the proband compared to the normal controls, which is different from previous cases.
- It is of interest that all patients with c.1901T>C mutation were late-onset type and selectively from Asian countries, especially Japan and China.
INVITED COMMENTARY
Indeterminate thyroid nodules in the era of molecular genomics
- First Published: 21 May 2020
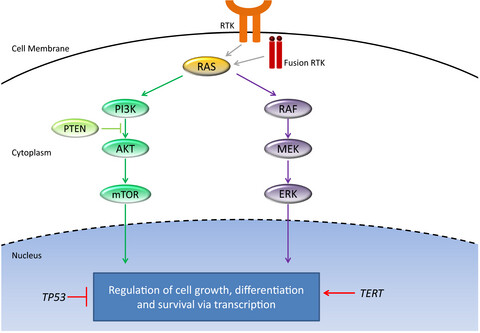
Indeterminate thyroid nodules are diagnosed in up to 30% of fine-needle aspirations and the risk of malignancy in these cases are highly variable. The introduction of molecular testing of these nodules provided a platform to help distinguish benign versus malignant nodules, though each test varies in predictability.
LETTER TO THE EDITOR
An audiological perspective on ‘‘Two further patients with Warsaw breakage syndrome. Is a mild phenotype possible?”
- First Published: 05 June 2020




