

Journal list menu

 Issue2019i-iii, 2167-2444
Issue2019i-iii, 2167-2444
Export Citations
Download PDFs
Front Cover, Volume 40, Issue 12
- Page: i
- First Published: 23 November 2019

Front Cover: The cover image is based on the Review Genome-wide transcriptomic and proteomic studies of Retisyndrome mouse models identify common signaling pathways and cellular functions as potential therapeutic targets by Rahul Krishnaraj et al., https://doi.org/10.1002/humu.23887.
Cover image © Florencia Haase Images.
Inside Back Cover, Volume 40, Issue 12
- Page: ii
- First Published: 23 November 2019

Inside Back Cover: The cover image is based on the Research Article CAPN5 genetic inactivation phenotype supports therapeutic inhibition trials by Katherine J. Wert et al., https://doi.org/10.1002/humu.23894.
Cover image © Katherine J. Wert, Gabriel Velez and Vinit B. Mahajan Images.
Back Cover, Volume 40, Issue 12
- Page: iii
- First Published: 23 November 2019
Back Cover: The cover image is based on the Research Article Alagille syndrome mutation update: Comprehensive overview of JAG1 and NOTCH2 mutation frequencies and insight into missense variant classification by Melissa A. Gilbert et al., https://doi.org/10.1002/humu.23879.
Cover image © Melissa Gillbert and Nancy Spinner Image.
Issue Information
- Pages: 2167-2169
- First Published: 23 November 2019
Single-nucleotide editing: From principle, optimization to application
- Pages: 2171-2183
- First Published: 27 May 2019
Genome-wide transcriptomic and proteomic studies of Rett syndrome mouse models identify common signaling pathways and cellular functions as potential therapeutic targets
- Pages: 2184-2196
- First Published: 05 August 2019
Alagille syndrome mutation update: Comprehensive overview of JAG1 and NOTCH2 mutation frequencies and insight into missense variant classification
- Pages: 2197-2220
- First Published: 25 July 2019
LOVD–DASH: A comprehensive LOVD database coupled with diagnosis and an at-risk assessment system for hemoglobinopathies
- Pages: 2221-2229
- First Published: 09 July 2019
Dutch genome diagnostic laboratories accelerated and improved variant interpretation and increased accuracy by sharing data
- Pages: 2230-2238
- First Published: 21 August 2019
Pitfalls in the interpretation of CFTR variants in the context of incidental findings
- Pages: 2239-2246
- First Published: 27 July 2019
Novel missense mutation in VPS33B is associated with isolated low gamma-glutamyltransferase cholestasis: Attenuated, incomplete phenotype of arthrogryposis, renal dysfunction, and cholestasis syndrome
- Pages: 2247-2257
- First Published: 03 September 2019
Novel ACTN1 variants in cases of thrombocytopenia
- Pages: 2258-2269
- First Published: 25 June 2019
Deleterious de novo variants of X-linked ZC4H2 in females cause a variable phenotype with neurogenic arthrogryposis multiplex congenita
- Pages: 2270-2285
- First Published: 17 June 2019
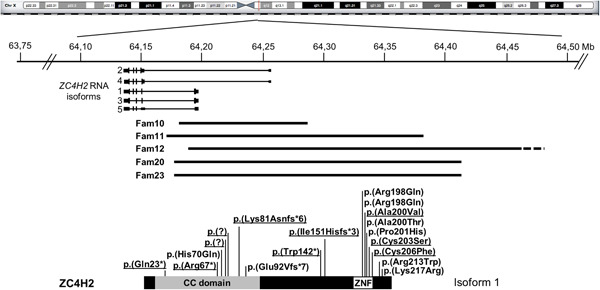
Overview of newly identified likely pathogenic variants in the X-linked gene ZC4H2 leading to ZC4H2-Associated Rare Disorders (ZARD) in affected males and females. The spectrum of ZC4H2 defects comprises novel and recurrent mostly inherited missense variants in affected males, and de novo splicing, frameshift, nonsense and partial ZC4H2 deletions in affected females.
Mutations in PLS1, encoding fimbrin, cause autosomal dominant nonsyndromic hearing loss
- Pages: 2286-2295
- First Published: 09 August 2019
Incorporation of semi-quantitative analysis of splicing alterations for the clinical interpretation of variants in BRCA1 and BRCA2 genes
- Pages: 2296-2317
- First Published: 25 July 2019
Genetical, clinical, and functional analysis of a large international cohort of patients with autosomal recessive congenital ichthyosis due to mutations in NIPAL4
- Pages: 2318-2333
- First Published: 26 July 2019
ATP1A1 mutations cause intermediate Charcot-Marie-Tooth disease
- Pages: 2334-2343
- First Published: 02 August 2019
Dominant-negative SOX9 mutations in campomelic dysplasia
- Pages: 2344-2352
- First Published: 07 August 2019
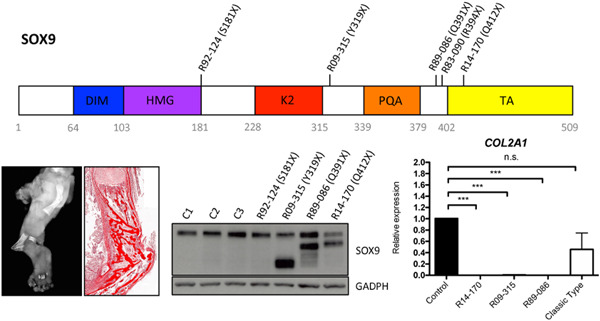
This study describes distal truncating mutations in SOX9, the chondrogenesis master regulator, in forms of campomelic dysplasia (CD), an autosomal dominant, perinatal lethal skeletal dysplasia. The mutations all leave the dimerization and DNA-binding domains intact and the truncated SOX9 exerted a dominant-negative effect, exemplified by its effect on COL2A1 expression. The data identify a novel molecular mechanism of disease in CD with a distinct and more significant effect on SOX9 function.
Expression and functional characterization of missense mutations in ATP8A2 linked to severe neurological disorders
- Pages: 2353-2364
- First Published: 09 August 2019
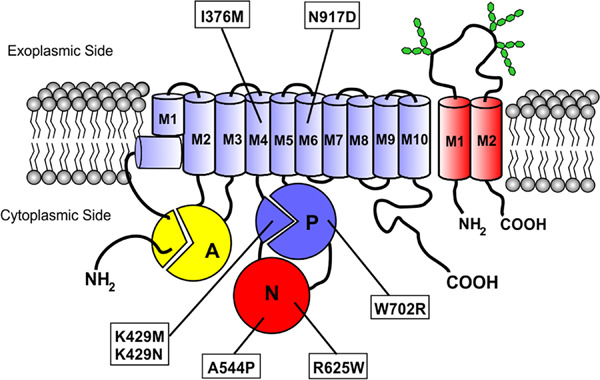
Mutations in the ATP8A2 gene encoding the phospholipid flippase ATP8A2 cause severe autosomal recessive neurological diseases in humans characterized by intellectual disability, hypotonia, chorea, hyperkinetic movement disorders, and optic and cerebellar atrophy. In the current study we examined the effect of missense mutations in the transmembrane and catalytic domains on the expression and functional activity of ATP8A2. All variants were devoid of functional activity. Three variants expressed at levels similar to normal ATP8A2 but lacked key residues required for function; four variants expressed at very low levels due to severe protein misfolding and proteosomal degradation.
Identification of splice defects due to noncanonical splice site or deep-intronic variants in ABCA4
- Pages: 2365-2376
- First Published: 09 August 2019
CAPN5 genetic inactivation phenotype supports therapeutic inhibition trials
- Pages: 2377-2392
- First Published: 12 August 2019
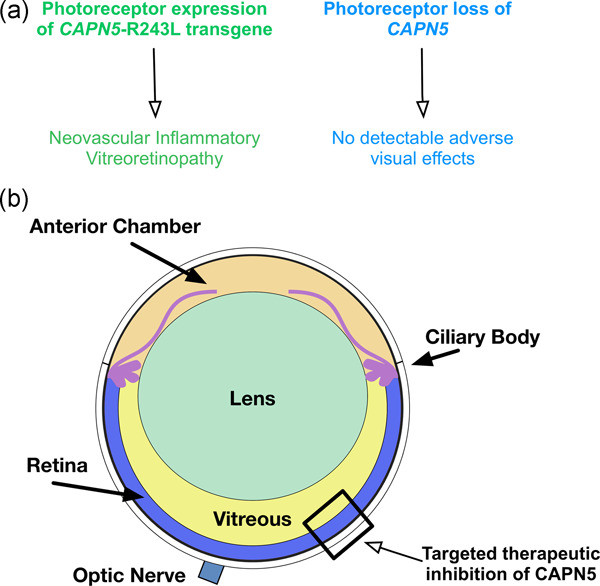
Small molecule pharmacological inhibition of dominant human genetic disease is a feasible treatment that does not rely on the development of individual, patient-specific gene therapy vectors. We created a mouse model with photoreceptor-specific loss of CAPN5 and examined it for any retinal abnormalities and disease presentation, as well as mining human databases for gene variants that cause a loss-of-function (LOF) in CAPN5. We not only found that tolerated, non-disease-causing human gene variants leading to LOF are present throughout CAPN5, even in regions thought to be highly susceptible to disease-causing mutations, but that knocking out CAPN5 within the retina does not cause any detectable retinal abnormalities, suggesting that targeted suppression of CAPN5 in the photoreceptor cells is a viable strategy and supports therapeutic inhibition trials.
De novo GRIN variants in NMDA receptor M2 channel pore-forming loop are associated with neurological diseases
- Pages: 2393-2413
- First Published: 20 August 2019
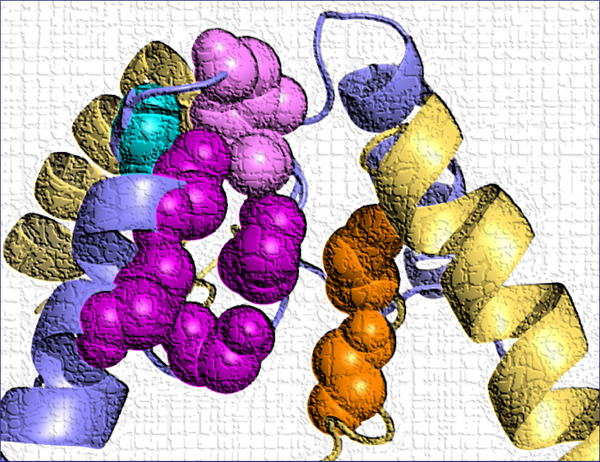
Disease-associated missense variants in M2 pore loop of GRIN genes
From incomplete penetrance with normal telomere length to severe disease and telomere shortening in a family with monoallelic and biallelic PARN pathogenic variants
- Pages: 2414-2429
- First Published: 26 August 2019
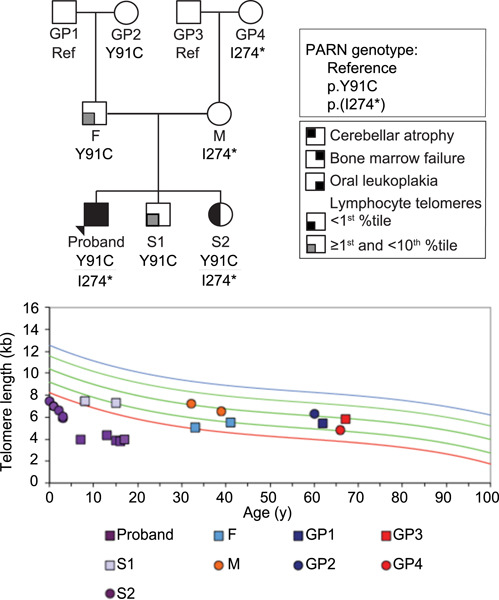
Heterozygous pathogenic variants in PARN, poly(A)-specific ribonuclease, are a cause of idiopathic pulmonary fibrosis, but with incomplete penetrance, whereas biallelic variants are a cause of Hoyeraal-Hreidarsson syndrome. Here we investigate a three-generation family with two pathogenic PARN variants dispersed among mono- and biallelic carriers. Comprehensive analysis of telomere length, PARN protein and RNA, and PARN RNA targets in every individual in the family reveals marked variability of effects among individuals.
Deciphering exome sequencing data: Bringing mitochondrial DNA variants to light
- Pages: 2430-2443
- First Published: 05 August 2019
Motor protein binding and mitochondrial transport are altered by pathogenic TUBB4A variants
- Page: 2444
- First Published: 04 November 2019