Journal list menu

 Issue
IssueClinical Genetics: Volume 99, Issue 3
333-489March 2021
Export Citations
Download PDFs
ISSUE INFORMATION
REVIEWS
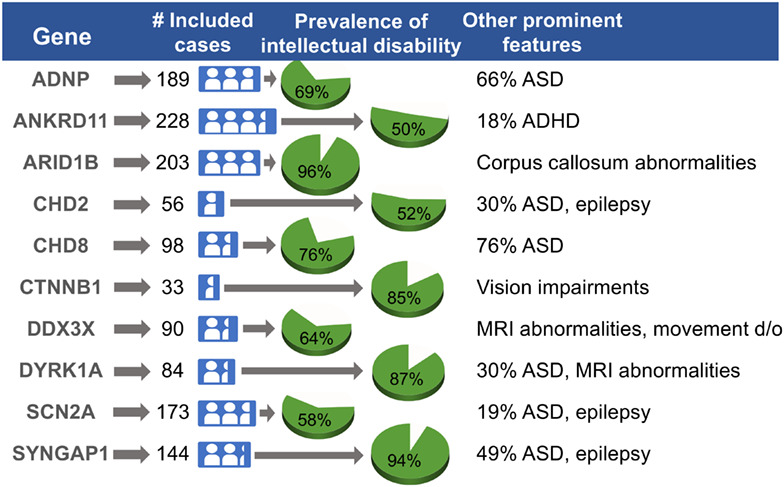
Description of neurodevelopmental phenotypes associated with 10 genetic neurodevelopmental disorders: A scoping review
- Pages: 335-346
- First Published: 11 November 2020
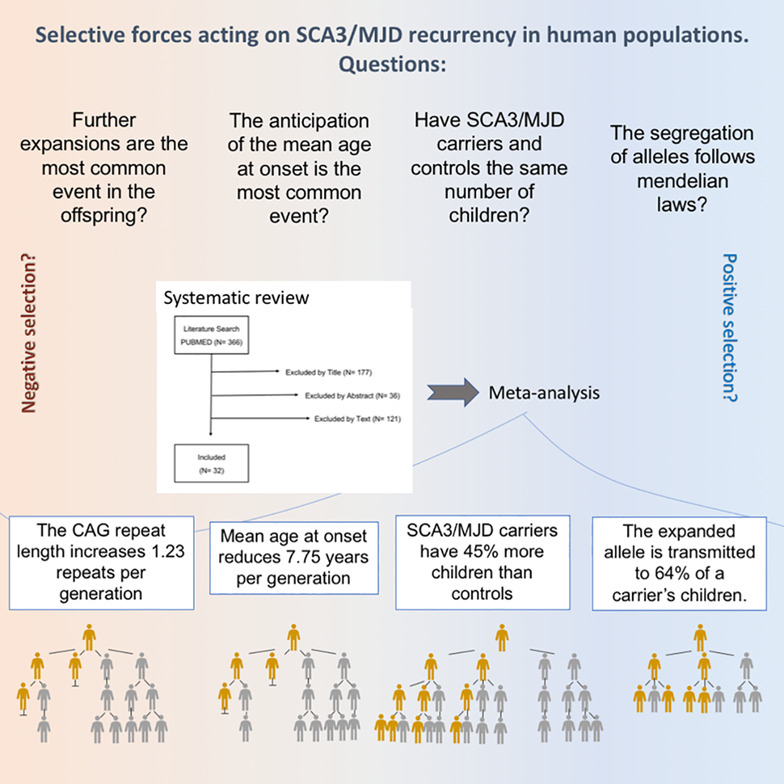
Selective forces acting on spinocerebellar ataxia type 3/Machado–Joseph disease recurrency: A systematic review and meta-analysis
- Pages: 347-358
- First Published: 21 November 2020
ORIGINAL ARTICLES
Genetic spectrum of Charcot–Marie–Tooth disease associated with myelin protein zero gene variants in Japan
- Pages: 359-375
- First Published: 11 November 2020
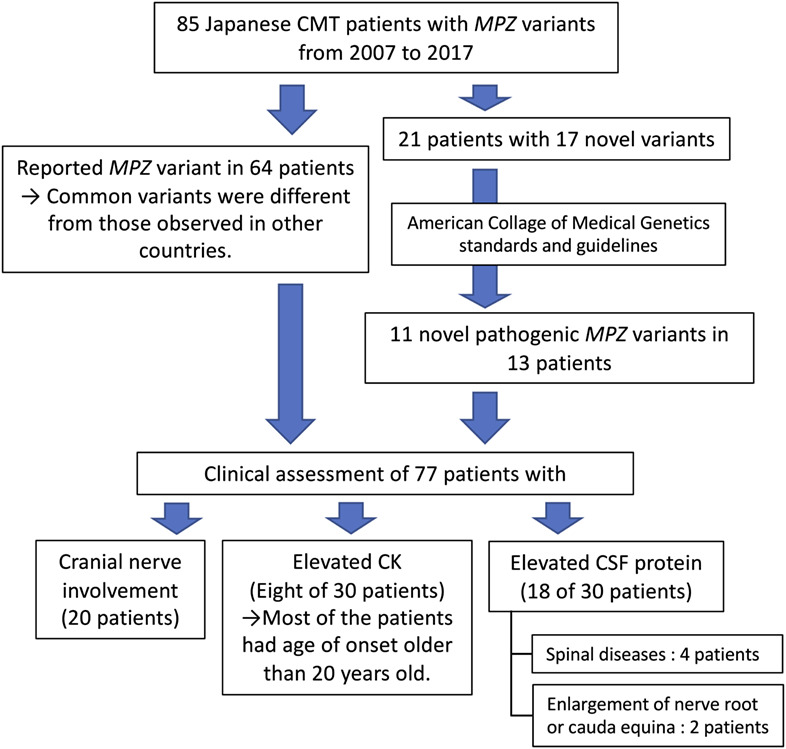
Genetic spectrum of MPZ variants was confirmed in this study.
Deficiency of acyl-CoA synthetase 5 is associated with a severe and treatable failure to thrive of neonatal onset
- Pages: 376-383
- First Published: 15 November 2020
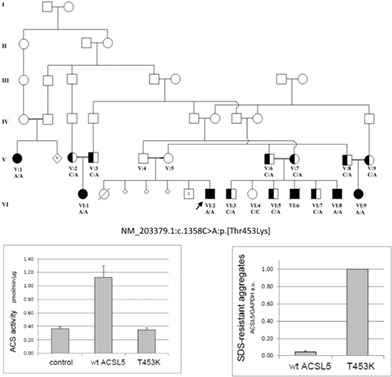
ACSL5 biallelic loss-of-function variants lead to severe failure to thrive, but treatable with dietary restriction of long-chain triglycerides.
Genetic variations and clinical spectrum of dystroglycanopathy in a large cohort of Chinese patients
- Pages: 384-395
- First Published: 16 November 2020
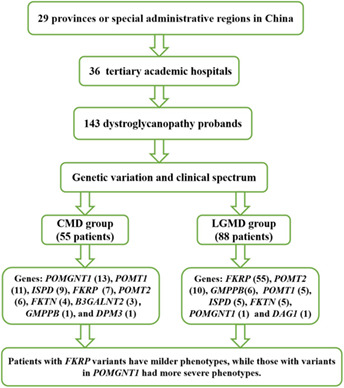
Null variants in DYSF result in earlier symptom onset
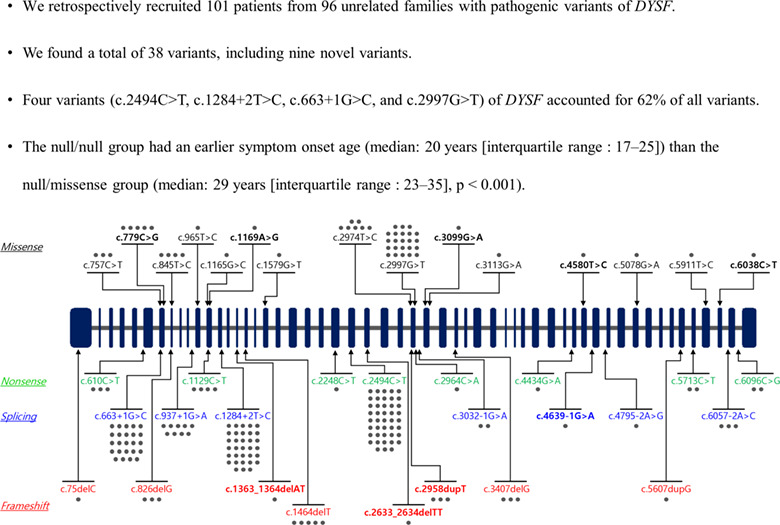
- Pages: 396-406
- First Published: 20 November 2020
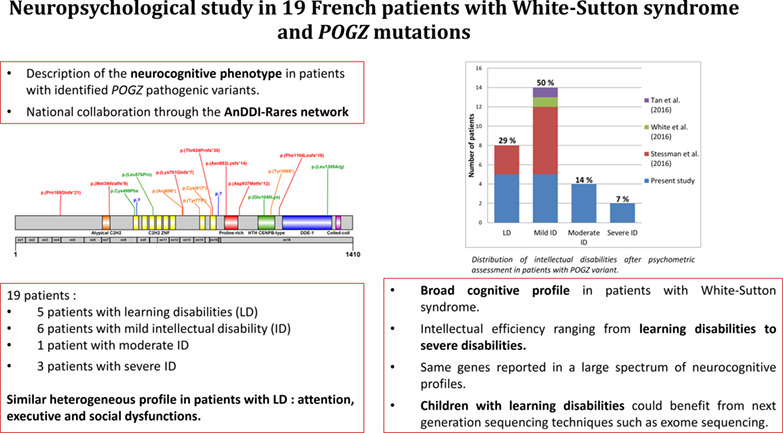
Neuropsychological study in 19 French patients with White-Sutton syndrome and POGZ mutations
- Pages: 407-417
- First Published: 05 December 2020
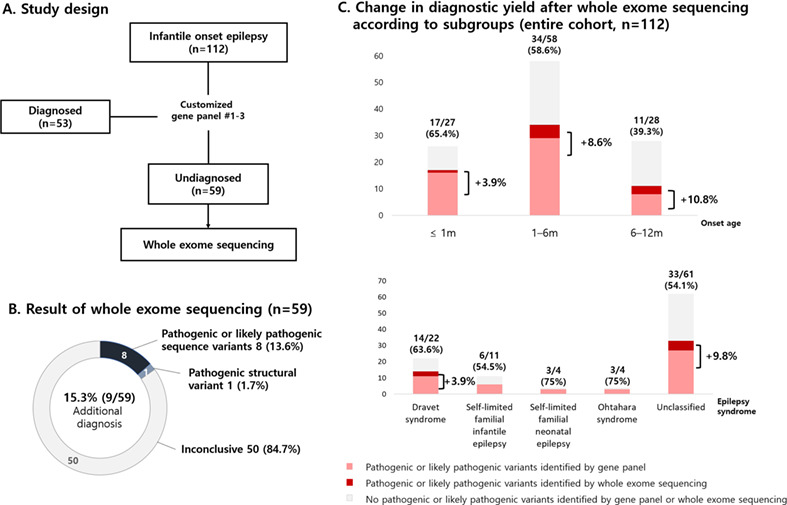
Genetic diagnosis of infantile-onset epilepsy in the clinic: Application of whole-exome sequencing following epilepsy gene panel testing
- Pages: 418-424
- First Published: 21 December 2020
SHORT REPORTS
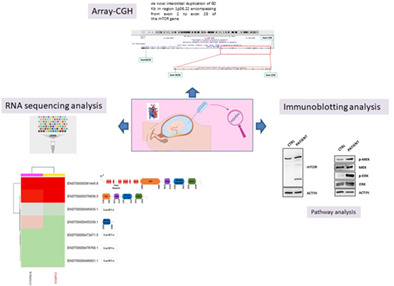
Functional evidence of mTORβ splice variant involvement in the pathogenesis of congenital heart defects
- Pages: 425-429
- First Published: 24 November 2020
Novel ACTG2 variants disclose allelic heterogeneity and bi-allelic inheritance in pediatric chronic intestinal pseudo-obstruction
- Pages: 430-436
- First Published: 08 December 2020
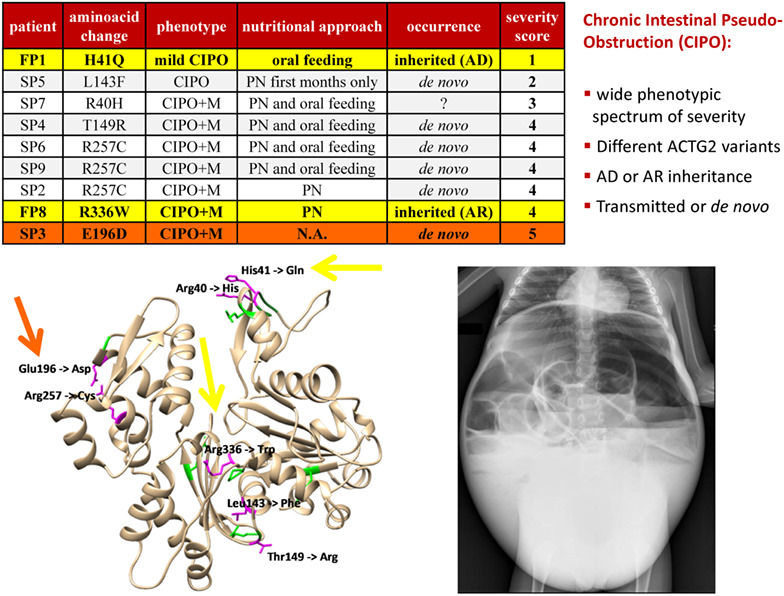
Chronic Intestinal Pseudo-Obstruction (CIPO):
- wide phenotypic spectrum of severity
- Different ACTG2 variants
- AD or AR inheritance
- Transmitted or de novo
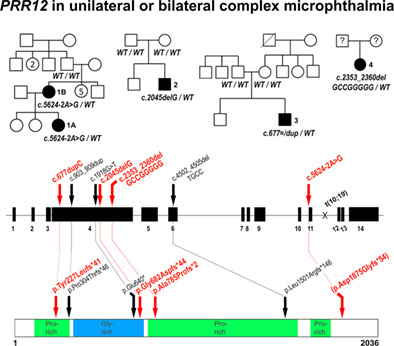
Dominant variants in PRR12 result in unilateral or bilateral complex microphthalmia
- Pages: 437-442
- First Published: 13 December 2020
Biallelic mutations of CFAP58 are associated with multiple morphological abnormalities of the sperm flagella
- Pages: 443-448
- First Published: 13 December 2020
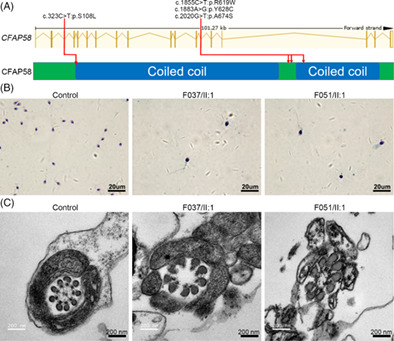
The multiple morphological abnormalities of the sperm flagella (MMAF) is one of the most severe types of teratozoospermia. In this study, we conducted whole-exome sequencing on 55 MMAF patients to identify the genetic etiology. Biallelic mutations of CFAP58 were identified from two patients. The variants are rare and disease causing, and CFAP58 was absent in the patients' sperm. Moreover, the TEM results showed the absence of central pairs and disruption of the DMTs. Intracytoplasmic sperm injection (ICSI) was carried out for couple of F037/II:1, his wife became pregnant and gave birth to a baby. This study further demonstrates that CFAP58 is a pathogenic gene of MMAF.
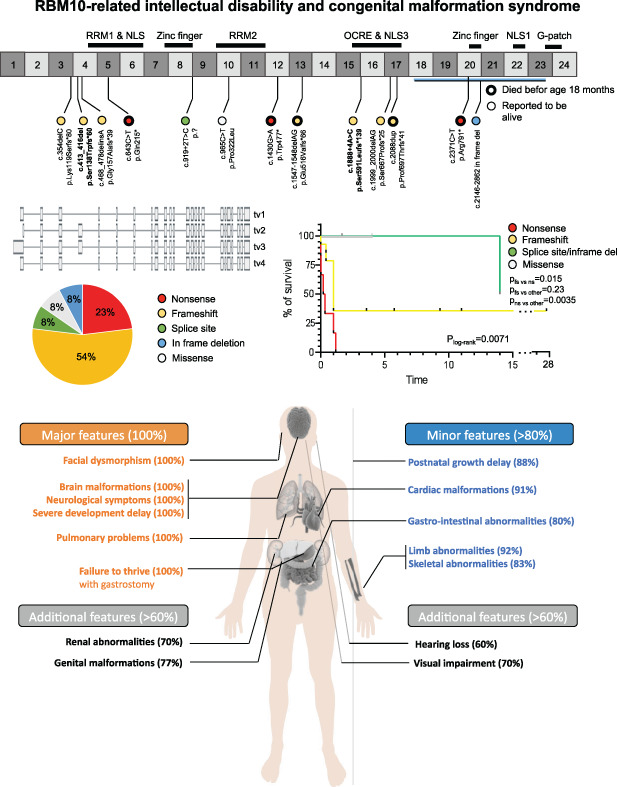
Phenotypic spectrum of the RBM10-mediated intellectual disability and congenital malformation syndrome beyond classic TARP syndrome features
- Pages: 449-456
- First Published: 19 December 2020
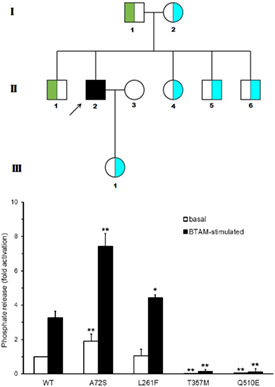
Compound heterozygosity for PTPN11 variants in a subject with Noonan syndrome provides insights into the mechanism of SHP2-related disorders
- Pages: 457-461
- First Published: 22 December 2020
IQSEC2 disorder: A new disease entity or a Rett spectrum continuum?
- Pages: 462-474
- First Published: 28 December 2020
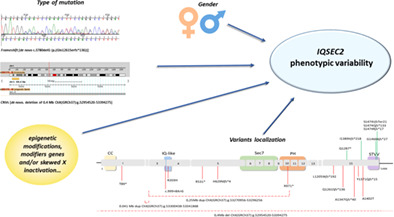
IQSEC2 mutations are associated to a neurodevelopment disorder (OMIM #300522) which shares many characteristics with Rett syndrome. With the aim of establishing a genotype-phenotype correlation, we collected molecular and clinical data of 16 patients harbouring IQSEC2 point mutations (15 previously unreported) and of 5 novel patients carrying CNVs encompassing IQSEC2, some of them being familial cases. Our data support the idea that position, type of variant and gender are crucial to determine the phenotype associated to IQSEC2 mutations.
LETTERS TO THE EDITOR
Psychosis in NUS1 de novo mutation: New phenotypical presentation
- Pages: 475-476
- First Published: 28 October 2020
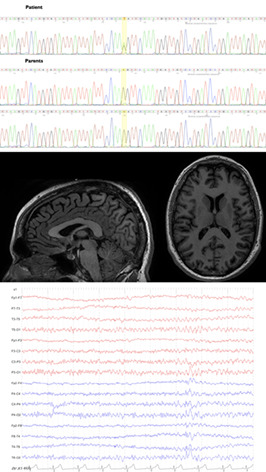
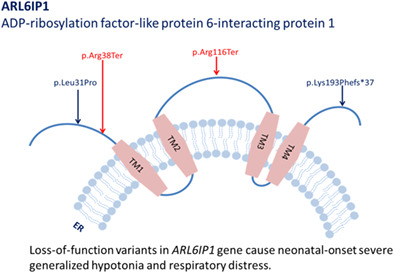
Genotype-phenotype study and expansion of ARL6IP1-related complicated hereditary spastic paraplegia
- Pages: 477-480
- First Published: 13 November 2020
Molecular and histologic insights on early onset cardiomyopathy in Danon disease females
- Pages: 481-483
- First Published: 23 November 2020

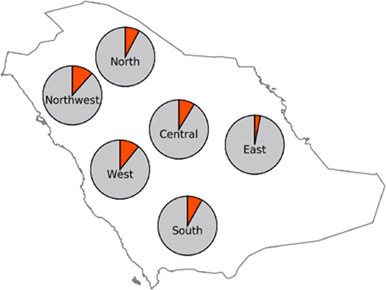
Indigenous Arabs have an intermediate frequency of a Neanderthal-derived COVID-19 risk haplotype compared with other world populations
- Pages: 484-485
- First Published: 27 November 2020
A novel stop codon variant affecting ΔNp63 isoforms associated with non-syndromic limb-mammary phenotype and uterine cervix dysplasia
- Pages: 486-487
- First Published: 01 December 2020
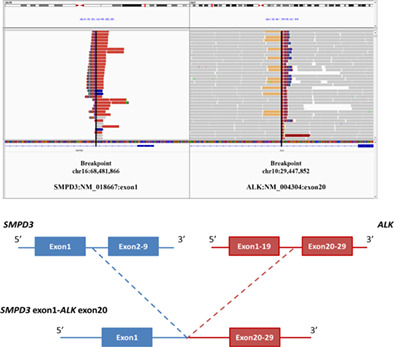
SMPD3-ALK: A novel ALK fusion gene in lung adenocarcinoma
- Pages: 488-489
- First Published: 08 February 2021