

Journal list menu

 Issue2018i-ii, 1151-1299
Issue2018i-ii, 1151-1299
Export Citations
Download PDFs
Cover Image, Volume 39, Issue 9
- Page: i
- First Published: 31 August 2018
On the Front Cover: This cover image, by Richard T. Wang et al., is based on the Research Article DMD genotype correlations from the Duchenne Registry: Endogenous exon skipping is a factor in prolonged ambulation for individuals with a defined mutation subtype, Pages 1193–1202. DOI: 10.1002/humu.23561.
Back Cover, Volume 39, Issue 9
- Page: ii
- First Published: 31 August 2018
On the Back Cover: This cover image, by Gemma Montalban et al., is based on the Brief Report Characterization of spliceogenic variants located in regions linked to high levels of alternative splicing: BRCA2 c.7976+5G > T as a case study, Pages 1155–1160. DOI: 10.1002/humu.23583. Cover Funding: Grant/Award Number: PI15/00355. Spanish Instituto de Salud Carlos III (ISCIII) funding, an initiative of the Spanish Ministry of Economy and Innovation partially supported by European Regional Development FEDER Funds.
Issue Information
- Pages: 1151-1154
- First Published: 31 August 2018
Characterization of spliceogenic variants located in regions linked to high levels of alternative splicing: BRCA2 c.7976+5G > T as a case study
- Pages: 1155-1160
- First Published: 03 July 2018
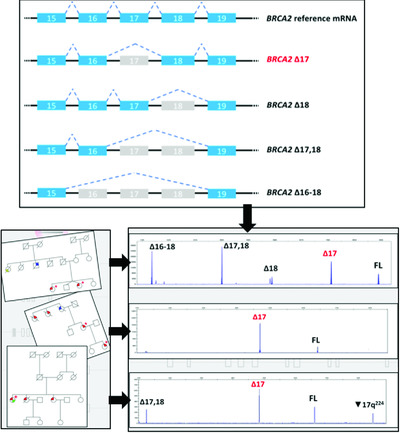
In this study we characterize BRCA2 c.7976 + 5G > T variant located in intron 17, which has an atypical donor site (GC). The variant promotes exon 17 skipping, but several isoforms (Δ16-18, Δ17,18, Δ18, and ▼17q224) have also been detected at different expression levels in variant carriers and controls. This study remarks the challenge of interpreting genetic variants located in regions with high levels of alternative splicing.
De novo mutations in FLNC leading to early-onset restrictive cardiomyopathy and congenital myopathy
- Pages: 1161-1172
- First Published: 01 June 2018
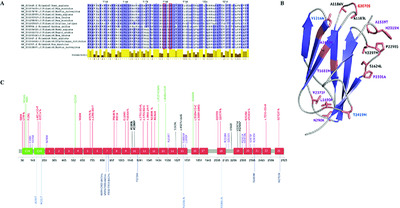
We report a new clinical phenotype of filaminopathy with early—onset restrictive cardiomyopathy in combination with congenital myopathy and arthrogryposisdue to de novoFLNC mutations. Described FLNC mutations in combination with functional studies extend cardiac spectrum of filaminopathies and facilitate the differential diagnosis of restrictive cardiac phenotype associated with neuromuscular involvement in children.
Results of next-generation sequencing gene panel diagnostics including copy-number variation analysis in 810 patients suspected of heritable thoracic aortic disorders
- Pages: 1173-1192
- First Published: 16 June 2018
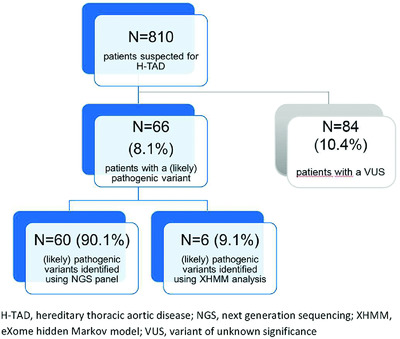
The aim of this study was to assess the diagnostic yield and the prevalence of copy number variants in patients suspected of hereditary thoracic aortic disease (H-TAD). A pathogenic or likely pathogenic variant was found in 66 of 810 patients (8.1%) and six out of these 66 likely pathogenic or pathogenic variants (9.1%) were copy number variants. Given the clinical relevance of identification of a genetic cause, copy number variant analysis should be incorporated into the clinical diagnostics of patients suspected of hereditary thoracic aortic disease.
DMD genotype correlations from the Duchenne Registry: Endogenous exon skipping is a factor in prolonged ambulation for individuals with a defined mutation subtype
- Pages: 1193-1202
- First Published: 16 June 2018
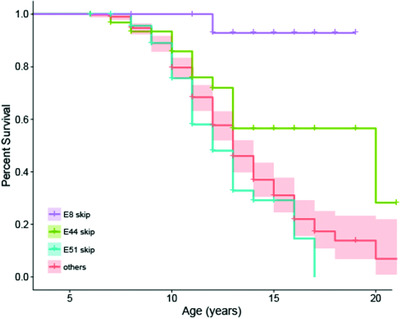
Using patient data from the Duchenne Registry, DMD individuals were stratified into separate groups according to the predicted exon skip necessary to produce an in-frame transcript. Delayed age at loss of ambulation was observed among DMD individuals amenable to skipping of exons 8 and 44, while exon 51 skippable individuals had earlier age of loss of ambulation.
Real-world clinical applicability of pathogenicity predictors assessed on SERPINA1 mutations in alpha-1-antitrypsin deficiency
- Pages: 1203-1213
- First Published: 07 June 2018
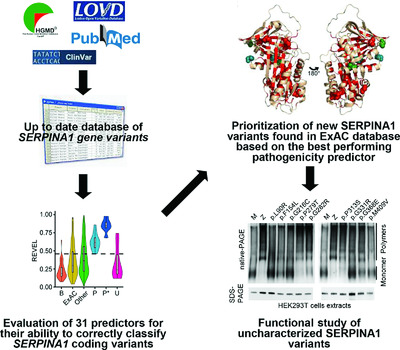
The growth of publicly available data informing upon genetic variations offers great potential for personalised medicine. We assessed performance of known pathogenicity predictors using SERPINA1 mutations causing α1-antitrypsin deficiency as a model. New mutations identified in the ExAC database and predicted as pathogenic by the best performing algorithms were characterised in cellular models. Our results overall support the potential of computational methods to provide meaningful predictions for novel mutations and identify areas for further investigation.
Germline variation in the oxidative DNA repair genes NUDT1 and OGG1 is not associated with hereditary colorectal cancer or polyposis
- Pages: 1214-1225
- First Published: 13 June 2018
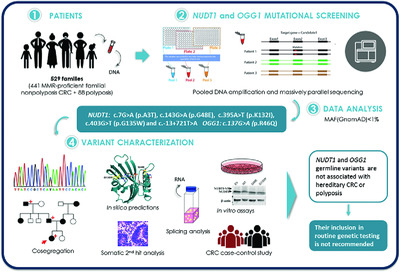
The evidence supporting a causal association of germline mutations in NUDT1 and OGG1 with hereditary colorectal cancer (CRC) or polyposis is currently insufficient/contradictory. We performed the mutational screening of the two genes in 529 families with unexplained mismatch repair-proficient familial nonpolyposis CRC or polyposis, followed by co-segregation analyses and extensive functional assays. Heterozygous NUDT1 and OGG1 germline variants are not a relevant cause of hereditary CRC or polyposis, therefore, their screening is not recommended in routine genetic testing.
Further delineation of Malan syndrome
- Pages: 1226-1237
- First Published: 13 June 2018
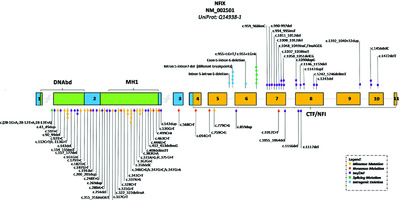
Malan is an overgrowth syndrome caused by NFIX variants, likely as frequent as other overgrowth syndromes. The overgrowth is present at birth but no longer evident in half of the adults. Facial characteristics are different from Sotos syndrome, clinical evaluation in itself allows differentiation of the two entities, and the designation “Sotos type 2” is no longer acceptable. NFIX variants can also cause Marshall-Smith syndrome, but with a different site of the stop codon, which explains the differences in phenotypes.
Targeted resequencing reveals genetic risks in patients with sporadic idiopathic pulmonary fibrosis
- Pages: 1238-1245
- First Published: 19 June 2018
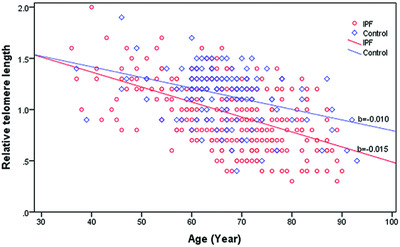
A total of 92 potentially IPF-related genes were selected and sequenced in genomic DNAs from 253 sporadic IPF patients and 125 matched health controls. We not only identified multiple novel rare and common variants that are associated with increased risk of IPF, but also constructed a cumulative risk model that suggests the possibility of risk prediction and stratification for IPF in Chinese.
Elucidating the genetic architecture of Adams–Oliver syndrome in a large European cohort
- Pages: 1246-1261
- First Published: 20 June 2018
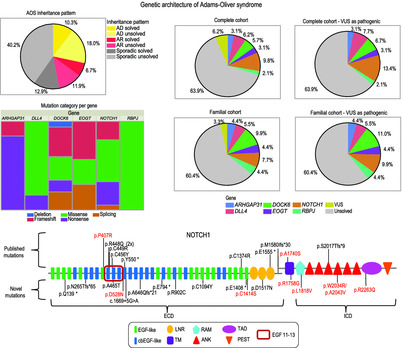
Adams–Oliver syndrome (AOS) is a rare developmental disorder, characterized by scalp aplasia cutis congenita (ACC) and transverse terminal limb defects (TTLD). Molecular diagnostic screening of 194 AOS/ACC/TTLD probands/families was conducted. In total, we identified 63 (likely) pathogenic mutations, providing a molecular diagnosis in 30% of patients. NOTCH1 is the major contributor, underlying 10% of AOS/ACC/TTLD cases, with DLL4 (6%), DOCK6 (6%), ARHGAP31 (3%), EOGT (3%), and RBPJ (2%) representing additional causality in this cohort.
Detailed analysis of HTT repeat elements in human blood using targeted amplification-free long-read sequencing
- Pages: 1262-1272
- First Published: 22 June 2018
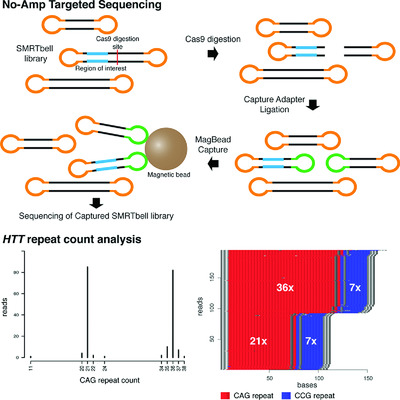
We have applied an amplification-free targeted sequencing protocol utilizing CRISPR/Cas9 (No-Amp Targeted sequencing) to study repeat elements in the huntingtin (HTT) gene, where an expanded CAG repeat is causative for Huntington disease. Eleven clinical samples were investigated, and for all alleles the resulting CAG repeat count agreed with previous results based on fragment analysis. With No-Amp Targeted sequencing, we can accurately sequence through repeat expansions that are difficult to investigate using PCR-based methods while also studying somatic variability between cells.
The analysis of myotonia congenita mutations discloses functional clusters of amino acids within the CBS2 domain and the C-terminal peptide of the ClC-1 channel
- Pages: 1273-1283
- First Published: 23 June 2018
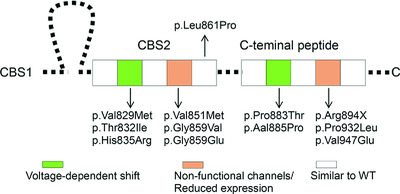
We provide clinical and patch-clamp analysis of ClC-1 chloride channel mutations causing Myotonia Congenita and located in the poorly characterized cytosolic C-terminal domain. The dysfunctions of mutant channels well correlated with the phenotypes. Clusters of ClC-1 mutations within CBS2 and C-terminal peptide sub-domains appeared to share similar functional defect. This study improves our understanding of the mechanisms underlying MC and of the role of the C-terminal region in ClC-1 function, providing information to develop new antimyotonic drugs.
Targeted sequencing with expanded gene profile enables high diagnostic yield in non-5q-spinal muscular atrophies
- Pages: 1284-1298
- First Published: 02 June 2018
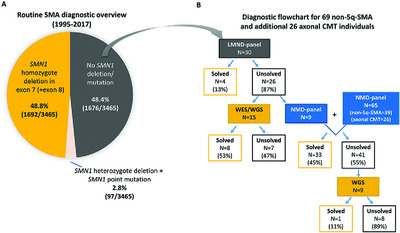
Of 3465 individuals with suspected spinal muscular atrophy (SMA) tested in routine diagnostic setting, about half failed to show an SMN1 deletion. To identify the genetic cause in these non-5q-SMA individuals, two gene panels: a small one with 62 lower motor neuron disease genes and a large one with 479 neuromuscular disease-related genes were developed. Unsolved cases were further analysed on WES/WGS. Based on our data, we discuss the best strategy for genetic testing of non-5q-SMA individuals.