

Journal list menu

 Issue2015i-v, 1021-1112, E2441-E2453
Issue2015i-v, 1021-1112, E2441-E2453
Export Citations
Download PDFs
Purifying Selection of mtDNA and the Shape of Human Evolution
- Page: v
- First Published: 12 October 2015
Neurofibromatosis Type 1 Without Neurofibromas: Genotype-Phenotype Correlations in NF1
- Page: v
- First Published: 12 October 2015
WDR73 Mutations Cause Infantile Neurodegeneration and Variable Glomerular Kidney Disease
- Pages: 1021-1028
- First Published: 29 June 2015
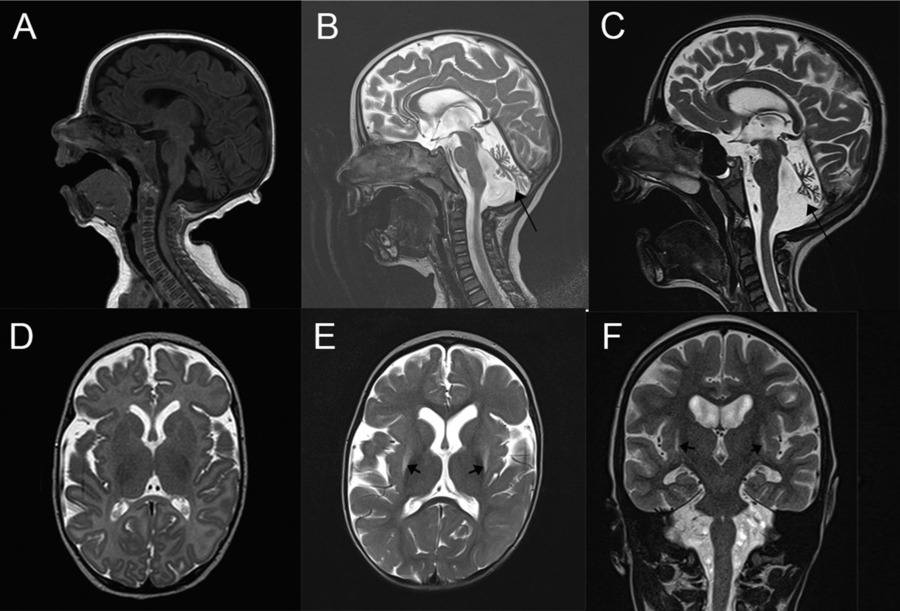
Homozygous WDR73 mutations result in infantile-onset neurodegeneration with striking cerebellar atrophy and basal ganglia alterations. Some patients also develop nephrotic syndrome.
Rare Variants in the Epithelial Cadherin Gene Underlying the Genetic Etiology of Nonsyndromic Cleft Lip with or without Cleft Palate
- Pages: 1029-1033
- First Published: 29 June 2015
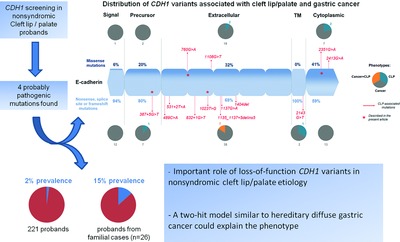
Rare, moderately penetrant variants in the epithelial cadherin gene consist in a promising risk factor for nonsyndromic cleft lip/palate. We highlight the importance of such variants in familial cases, and we suggest haploinsufficiency of CDH1 as the probable mechanism. To speculate why some CDH1 pathogenic variants may alternatively result in gastric cancer, we suggest a two-hit model underlying both phenotypes.
Complex Multiple-Nucleotide Substitution Mutations Causing Human Inherited Disease Reveal Novel Insights into the Action of Translesion Synthesis DNA Polymerases
- Pages: 1034-1038
- First Published: 14 July 2015
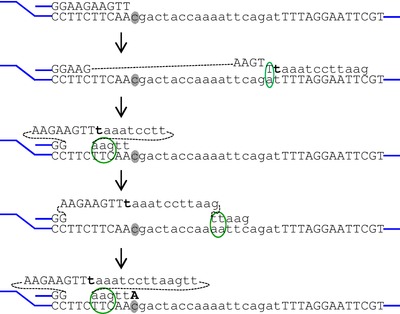
Translesion synthesis (TLS) DNA polymerases allow the bypass of unrepaired lesions during DNA replication. We have recently postulated new properties of TLS DNA polymerases in DNA repair: the generation of neo-microhomologies potentiating strand-misalignment and additional micro-lesions within the templated inserts when recruited to stalled replication forks. Here, we provide further support for this postulate and illustrate how repeated participation of TLS DNA polymerases could convert a damaged base into a complex lesion through a process of successive template switching and bypass repair.
Novel Homozygous Mutation of the Internal Translation Initiation Start Site of VHL is Exclusively Associated with Erythrocytosis: Indications for Distinct Functional Roles of von Hippel-Lindau Tumor Suppressor Isoforms
- Pages: 1039-1042
- First Published: 29 July 2015
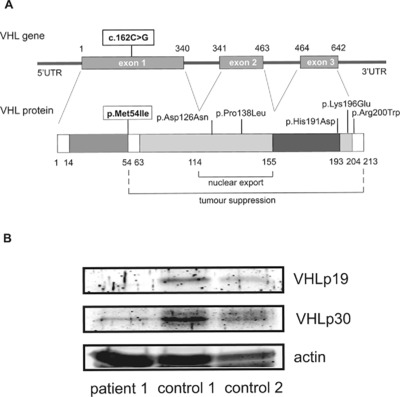
Mutations in VHL are either associated with the von Hippel Lindau tumor predisposition syndrome or congenital secondary erythrocytosis. In this report we describe three patients with erythrocytosis in whom we identified a unique mutation that completely abolishes production of one of the two VHL isoforms. Our findings in these patients point to differences in the functional roles of the two VHL isoforms. that is likely to have important implications for the fields of both erythropoiesis and tumorigenesis.
A New Homozygous IGF1R Variant Defines a Clinically Recognizable Incomplete Dominant form of SHORT Syndrome
- Pages: 1043-1047
- First Published: 07 August 2015
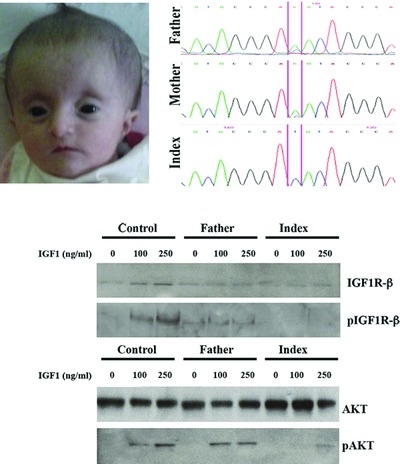
The proband presented with clinical features of SHORT syndrome, high IGF1 levels, developmental delay, CNS defects, and marked progeroid appearance. Anew homozygous c.2201G>T missense mutation in the IGF1R gene was identified. Functional studies in patient's cell lines showed a lower IGF1R expression that leads to the alteration of IGFlR-mediated AKT downstream pathway.
Expanding the Molecular and Clinical Phenotype of SSR4-CDG
- Pages: 1048-1051
- First Published: 12 August 2015
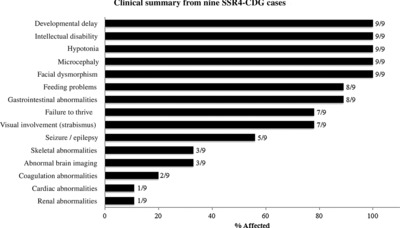
Congenital Disorders of Glycosylation (CDG) lead to incomplete protein glycosylation. One disorder, SSR4-CDG, was previously defined in a single patient. Here we present molecular analysis and clinical phenotypes of eight males with inherited and de novo mutations in X-linked SSR4. Standard CDG biochemical analysis might easily miss them.
High Incidence of Noonan Syndrome Features Including Short Stature and Pulmonic Stenosis in Patients carrying NF1 Missense Mutations Affecting p.Arg1809: Genotype–Phenotype Correlation
- Pages: 1052-1063
- First Published: 14 July 2015
FAS Gene Copy Numbers are Associated with Susceptibility to Behçet Disease and VKH Syndrome in Han Chinese
- Pages: 1064-1069
- First Published: 02 July 2015

The present study suggests that a high FAS gene copy number is associated with an increased risk of Behçet disease and VKH syndrome in Han Chinese, which is likely related to an increased expression of FAS in patients with Behçet disease and VKH syndrome.
POLD1 Germline Mutations in Patients Initially Diagnosed with Werner Syndrome
- Pages: 1070-1079
- First Published: 14 July 2015
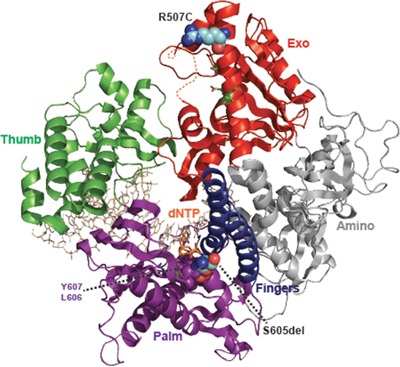
We have systematically analysed the POLD1 gene in 50 patients initially diagnosed with Werner Syndrome in whom no mutations in the Werner helicase gene were identified. Indeed, we found eight patients with heterozygous POLD1 mutations, thereby identifying the most common molecular cause of atypical Werner Syndrome until now. In addition, we could more than double the overall number of progeroid patients with POLD1 mutations, which allowed to substantially expand and modify the clinical characterization of this new example of a segmental progeroid disorder.
Activating Mutations Affecting the Dbl Homology Domain of SOS2 Cause Noonan Syndrome
- Pages: 1080-1087
- First Published: 14 July 2015

Noonan syndrome-causing mutations alter the Dbl homology domain (DH, shown as ribbon), altering its interaction with the RAS exchange motif, colored by its electrostatic potential. The mutated residue in the DH domain are colored red.
Concurrent DNA Copy-Number Alterations and Mutations in Genes Related to Maintenance of Genome Stability in Uninvolved Mammary Glandular Tissue from Breast Cancer Patients
- Pages: 1088-1099
- First Published: 29 July 2015
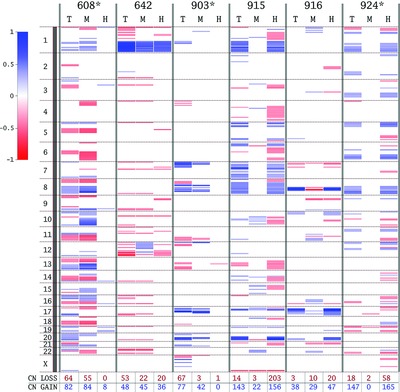
Panel A of the Figure 1 may be used for the graphical Table of Contents. The relevant high resolution TIF file is attached to the author proof. Here is the text of the proposed description: “Somatic mosaicism for DNA copy-number alterations (SMC-CNAs) is defined as gain or loss of chromosomal segments in somatic cells within a single organism. Here, gross SMC-CNAs (>500 kbp) were characterized in uninvolved mammary glandular tissue samples (6/59) from breast cancer patients, distal from the primary tumor site. These SMC-CNAs co-occurred with mutations and rare variants in genes related to maintenance of genomic integrity. Our results highlight the temporal and spatial neoplastic potential of uninvolved glandular tissue in breast cancer patients.”
Fine Time Scaling of Purifying Selection on Human Nonsynonymous mtDNA Mutations Based on the Worldwide Population Tree and Mother–Child Pairs
- Pages: 1100-1111
- First Published: 07 August 2015
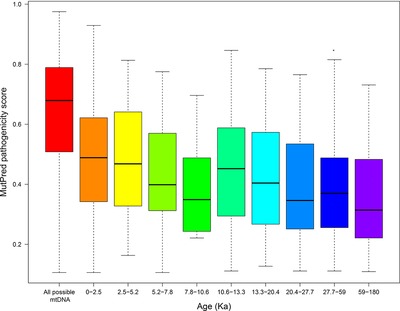
A high-resolution mtDNA phylogenetic tree allowed us to investigate purifying selection, which was very strong in the last 2,500 years. At the most recent time scale in 124 mother–children transmissions, purifying selection was detectable through the loss of mtDNA variants with high predicted pathogenicity. A high level of reversion of haplogroup-defining mutations was found in both the phylogenetic and single generation datasets, a proof of concept of a common selection dynamic upon mtDNA variation across so diverse time scales.
DOCK6 Mutations Are Responsible for a Distinct Autosomal-Recessive Variant of Adams–Oliver Syndrome Associated with Brain and Eye Anomalies
- Page: 1112
- First Published: 07 August 2015
Mediterranean Founder Mutation Database (MFMD): Taking Advantage from Founder Mutations in Genetics Diagnosis, Genetic Diversity and Migration History of the Mediterranean Population
- Pages: E2441-E2453
- First Published: 14 July 2015