

Journal list menu

 Issue
IssueJournal of Computational Chemistry: Volume 41, Issue 5
Special Issue:Membrane Protein Simulations and Free Energy Approaches: In honor of Professor Benoit Roux's 60th birthday
C1, 379-481February 15, 2020Guest Editor(s):
Export Citations
Download PDFs
Cover Image
Cover Image, Volume 41, Issue 5
- Page: C1
- First Published: 16 January 2020

I would like to express my gratitude to Wonpil Im, Nilesh Banavali, and Yun Luo and all the contributors for this Special Issue of the Journal of Computational Chemistry in celebration of my 60th birthday. To put it simply, I am very touched by the numerous contributions, and frankly humbled by the scope of the research presented here. Over the years, I had the pleasure and the good chance to cross path with many of these authors, students, postdocs, collaborators, getting a slice of the world as I was working with wonderfully bright people from all continents.
The more I think about this journey, and as Jacques Brel would put it, “Mes souvenir deviennent, ce que les vieux en font…” (Les Marquises 1977), good chance is certainly an undeniably important factor. Clearly, good chance was involved in meeting Rémy Sauvé who introduced me to the fascinating world of patch clamp and single channel recordings, Dennis Salahub who conveyed his passion for quantum chemistry, Martin Zuckermann who communicated his enthusiasm for statistical mechanics, and finally Martin Karplus my Ph.D. thesis advisor who will always be a constant inspiration. Let there be many more years of research and scientific investigations!
Issue Information
Editorial
Celebrating Benoît Roux's 60th birthday: quantifying biology at the membrane
- Pages: 385-386
- First Published: 16 January 2020
Review
Simulating ion channel activation mechanisms using swarms of trajectories
- Pages: 387-401
- First Published: 19 November 2019
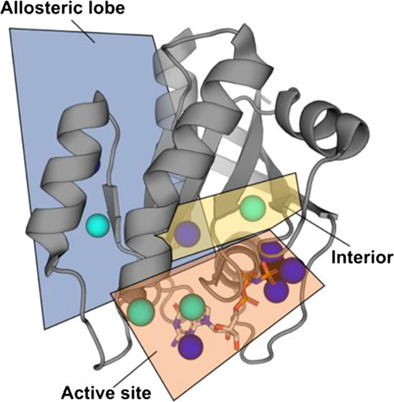
Studies of protein conformational change represent a significant challenge to computer simulations, owing to their wide-ranging length and time scales. Enhanced sampling approaches that can converge on the most likely path of change can help describe protein activity. This is illustrated with Roux's “swarms of trajectories” string method, which has been used to solve the allosteric gating mechanisms for a pentameric ligand-gated ion channel, yielding the molecular events, free energies, and kinetics to explain those hidden processes.
Full Papers
Water in Ras Superfamily Evolution
- Pages: 402-414
- First Published: 04 September 2019
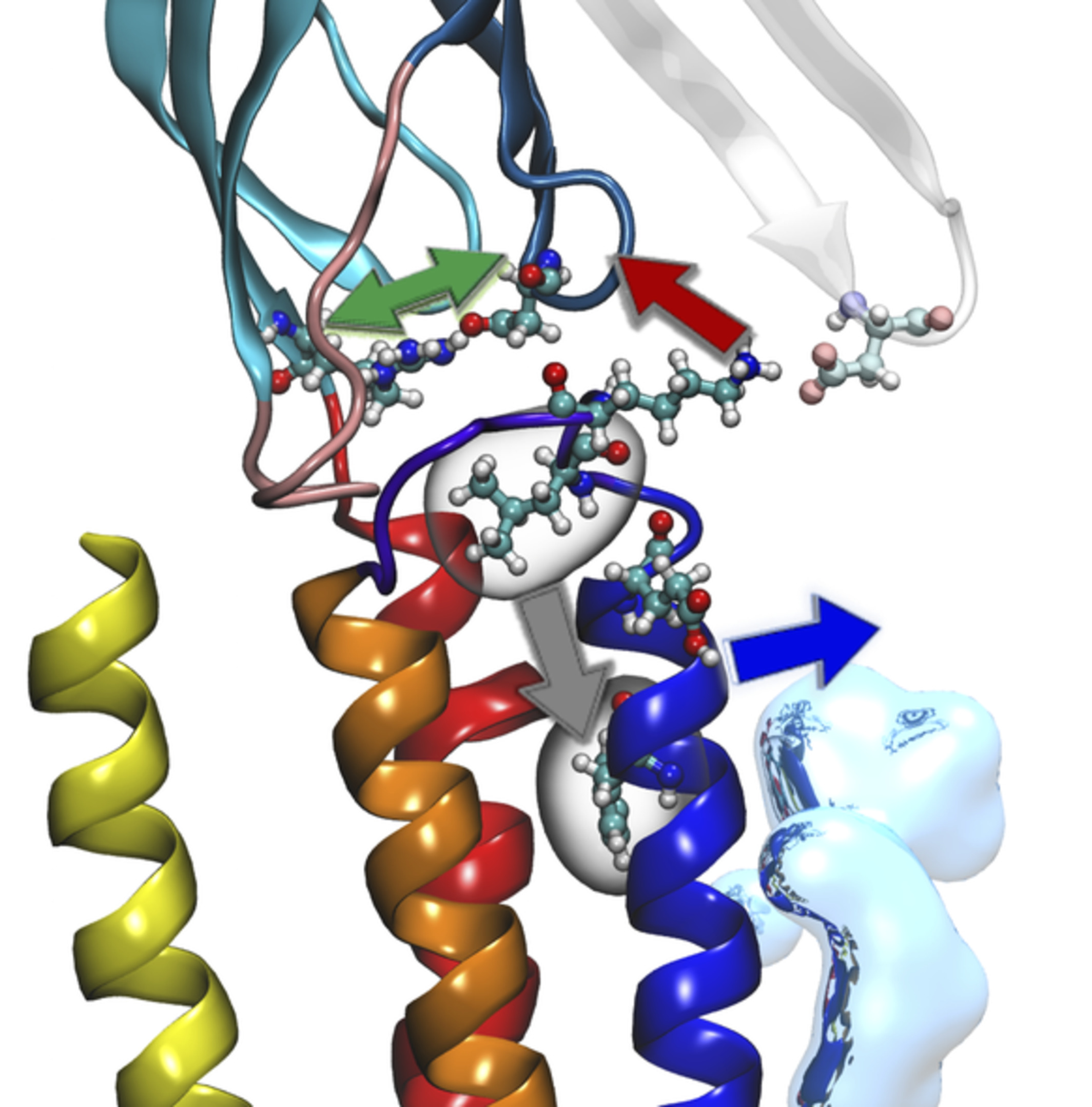
Given that the process of protein evolution occurs in an aqueous environment, the inclusion of water molecules in structural and functional adaptation is likely to be a major response to the selection pressures that drive evolution. Conserved water-binding sites retained across the Ras superfamily (shown in the figure) are associated with the common switch mechanism and serve as anchors for water-mediated networks that evolved separately in the Ras, Rho, Rab, and Arf branches.
CHARMM-GUI DEER facilitator for spin-pair distance distribution calculations and preparation of restrained-ensemble molecular dynamics simulations
- Pages: 415-420
- First Published: 22 July 2019

The double electron–electron resonance (DEER) is a powerful technique in structural biology to obtain distance information two unpaired electron spins. DEER Facilitator in CHARMM-GUIis presented as a new tool that consists of two modules Spin-Pair Distributor and reMD Prepper to setup molecular dynamics simulations that utilize information from DEER experiments.
Modulation of membrane permeability by carbon dioxide
- Pages: 421-426
- First Published: 03 September 2019
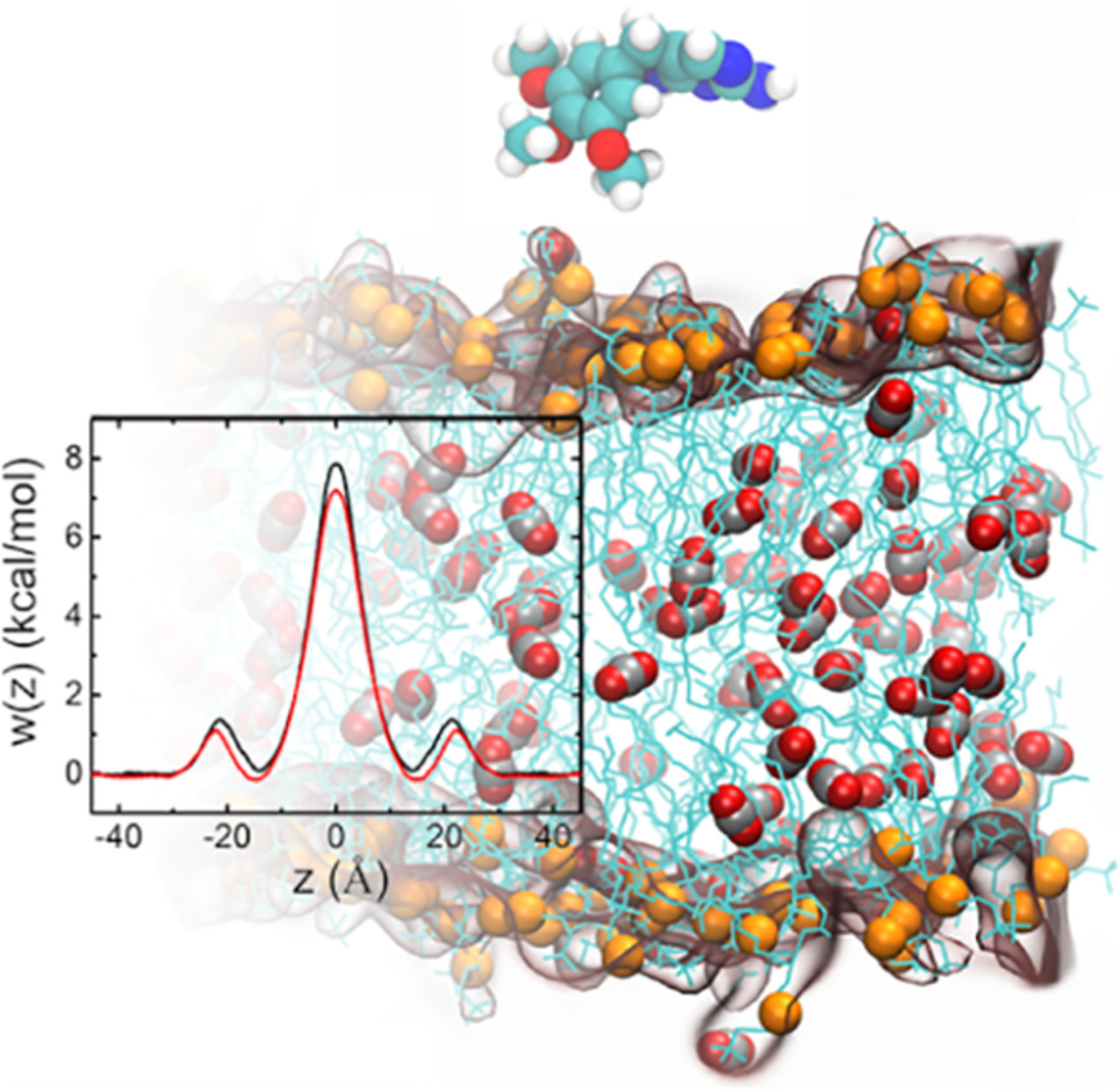
In the present work, modulation by CO2 of the membrane permeability to drugs has been investigated combining free energy and diffusivity calculations within the framework of the fractional solubility-diffusion model of permeation. The results indicate that CO2 loosens the membrane, thereby increasing the permeability to drugs. The present contribution provides a newfangled view that the thermodynamics and kinetics of permeation events can be modulated by gaseous species.
Quantum Chemical Methods for Modeling Covalent Modification of Biological Thiols
- Pages: 427-438
- First Published: 11 September 2019
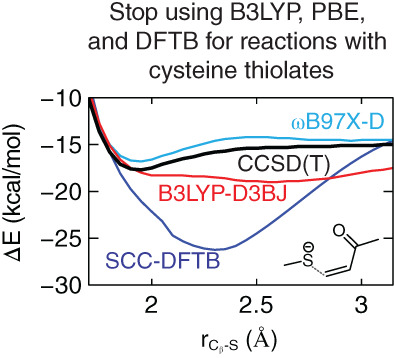
Reactions involving cysteine thiols often involve an anionic thiolate intermediate. Delocalization error in many popular density-functional theory (DFT) methods results in incorrect predictions of thio-Michael addition mechanisms of covalent-modifier drugs, while large exact-exchange components or range-separated functionals address these issues to some degree. Density-Functional Tight-Binding (DFTB) methods also fail to predict a stable enolate, while some semiempirical methods provide qualitative agreement with high-level theory. The thio-Michael additions are found to be an extremely stringent test of electronic structure methods, highlighting the need for accurate treatment of both electron delocalization and nonbonded repulsion.
Improved Modeling of Cation-π and Anion-Ring Interactions Using the Drude Polarizable Empirical Force Field for Proteins
- Pages: 439-448
- First Published: 13 September 2019
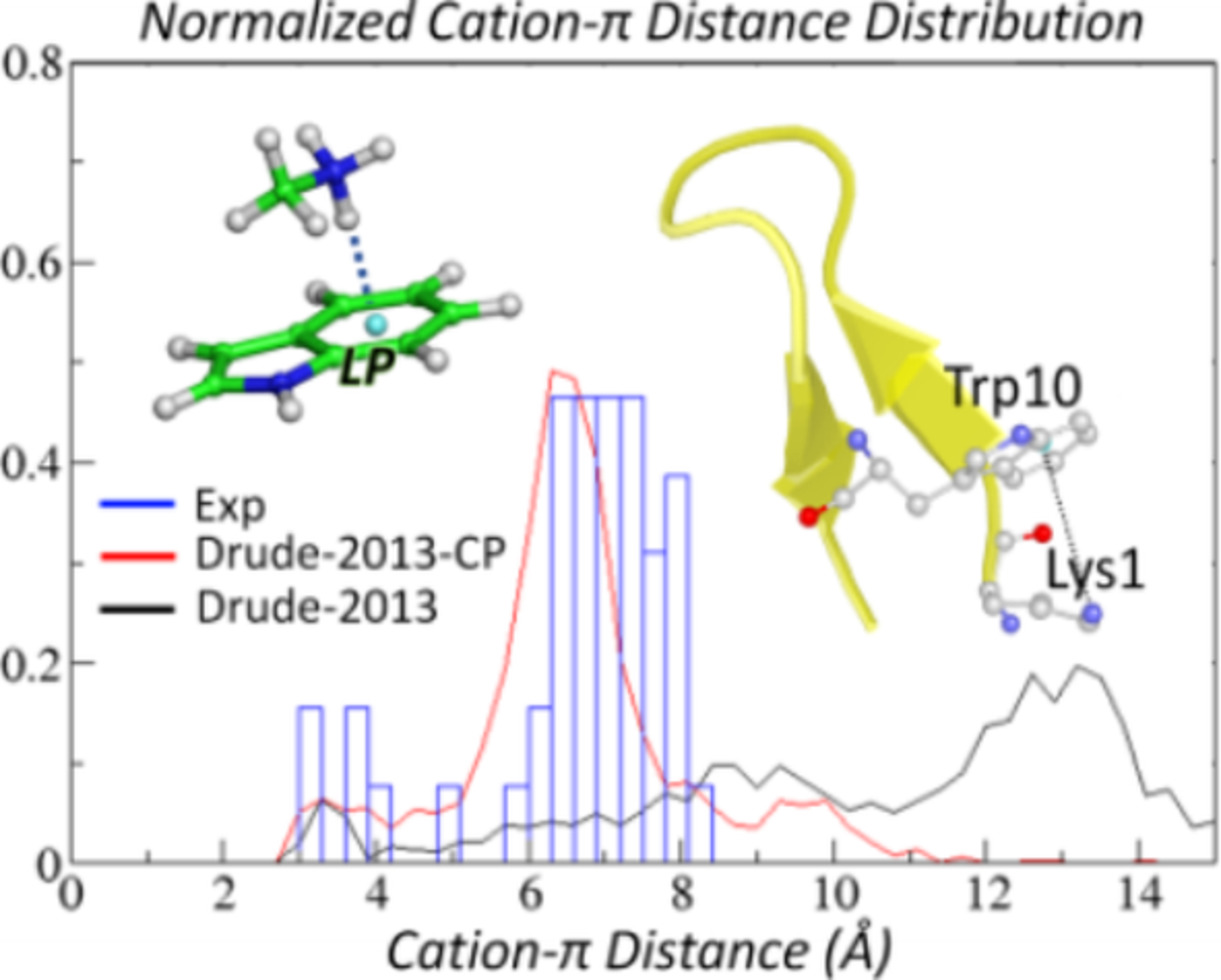
Cation-π and anion-ring interactions are common in proteins. To improve the accuracy of molecular dynamics simulations, the Drude-2013 polarizable protein force field was optimized targeting QM model compound interaction energies. The resulting force field, Drude-2013-CP, is demonstrated to be capable of reproducing experimentally identified cation-π and anion-ring interactions among multiple protein systems.
Direct Derivation of Free Energies of Membrane Deformation and Other Solvent Density Variations From Enhanced Sampling Molecular Dynamics
- Pages: 449-459
- First Published: 11 October 2019

A simulation methodology to calculate the potential of mean force of an arbitrary deformation of a lipid bilayer is presented. The membrane need not be homogeneous or symmetric, and lipids might be atomically detailed or coarse-grained. The central element is a grid-based collective variable denominated Multi-Map, which quantifies the membrane similarity to a set of density fields reflecting the transformation of interest. The proposed approach may be adapted to study other solvent reorganization processes.
G-Protein-Coupled Receptor–Membrane Interactions Depend on the Receptor Activation State
- Pages: 460-471
- First Published: 10 October 2019
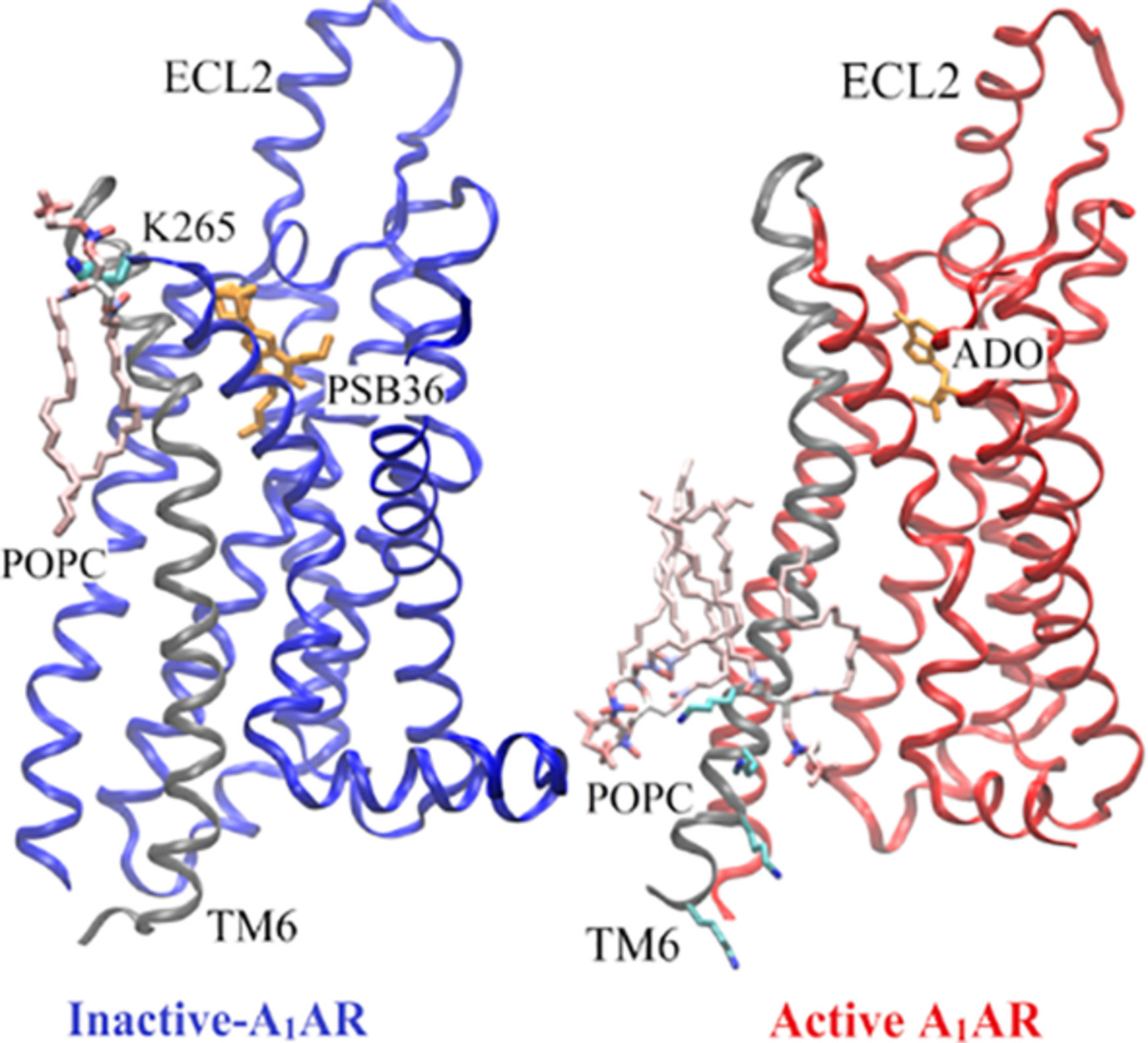
Using adenosine A1 receptor (A1AR) as a model G-protein-coupled receptor (GPCR), Gaussian accelerated molecular dynamics (GaMD) was applied to explore GPCR–membrane interactions. Membrane lipids played a key role in stabilizing different conformational states of the A1AR. Activation of the A1AR led to differential dynamics in the upper and lower leaflets of the lipid bilayer. The GaMD simulations revealed strongly coupled dynamics of the GPCR and lipids that depend on the receptor activation state.
Drude polarizable force field for cation–π interactions of alkali and quaternary ammonium ions with aromatic amino acid side chains
- Pages: 472-481
- First Published: 25 October 2019
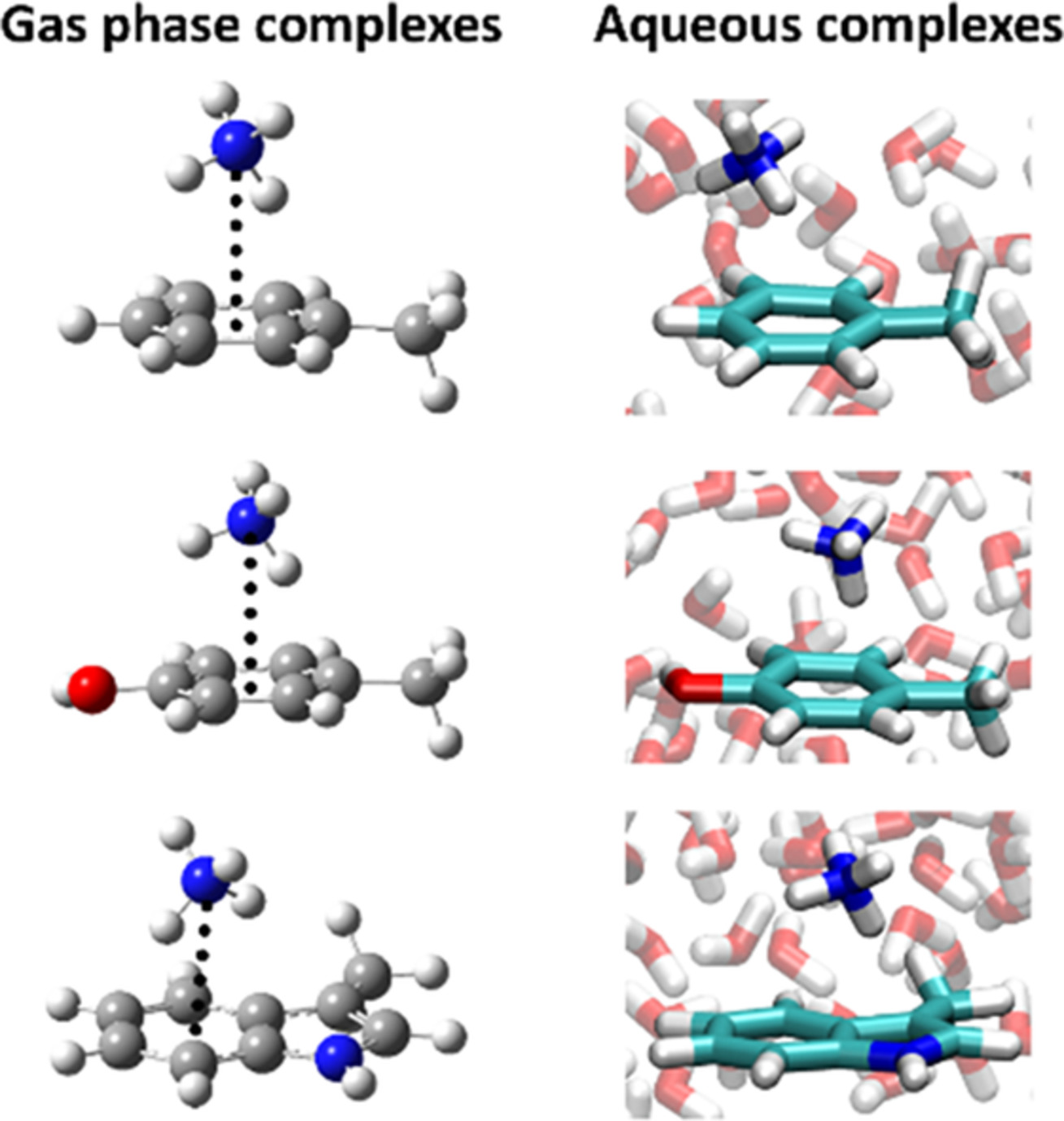
Cation–π interactions are important for protein stability and for molecular recognition in general, yet are not always adequately described by molecular mechanics models. The Drude polarizable force field is optimized for the interaction of Li+, Na+, K+, Rb+, Cs+, NH4+, (CH3)4+, and (C2H5)4N+ with aromatic compounds representing the side chains of Phe, Tyr, and Trp and used to evaluate the stability and geometry of the complexes in water.
Sign up for email alerts
Tools
