


The International Journal of Quantum Chemistry features an exciting mix of comprehensive reviews, instructive tutorials, visionary perspectives, and high-impact rapid communications and full papers that represent the entire field of quantum chemistry and molecular quantum mechanics, from theory to simulations and applications. A leading source of developments in quantum chemistry, we have published breakthroughs in theoretical and mathematical chemistry since 1967.
Journal Metrics
- 4.8CiteScore
- 2Journal Impact Factor
- 29%Acceptance rate
- 29 days Submission to first decision
International Journal of Quantum Chemistry is inviting applications for the role of Editor-in-Chief
We are excited to be seeking a new Editor-in-Chief to build on the journal’s strong foundations and continue to develop International Journal of Quantum Chemistry as a core community resource. The deadline for applications is September 30th 2025. Please click here for further details about the role and how to apply.
International Journal of Quantum Chemistry is inviting applications for the role of Associate Editor
As we invite applications for a new Editor-in-Chief, we are also excited to invite applications for Associate Editor roles. The deadline for applications is September 30th 2025. Please click here for further details about the Associate Editor role and how to apply.
On the Cover

[Correction added on 25 July 2024, after first online publication: Cover has been replaced.]
Articles
The Effect of Conjugation Length and NN Location on the Nonlinear Optical Switching Properties of Azobenzene Derivatives
- 19 July 2025
Graphical Abstract
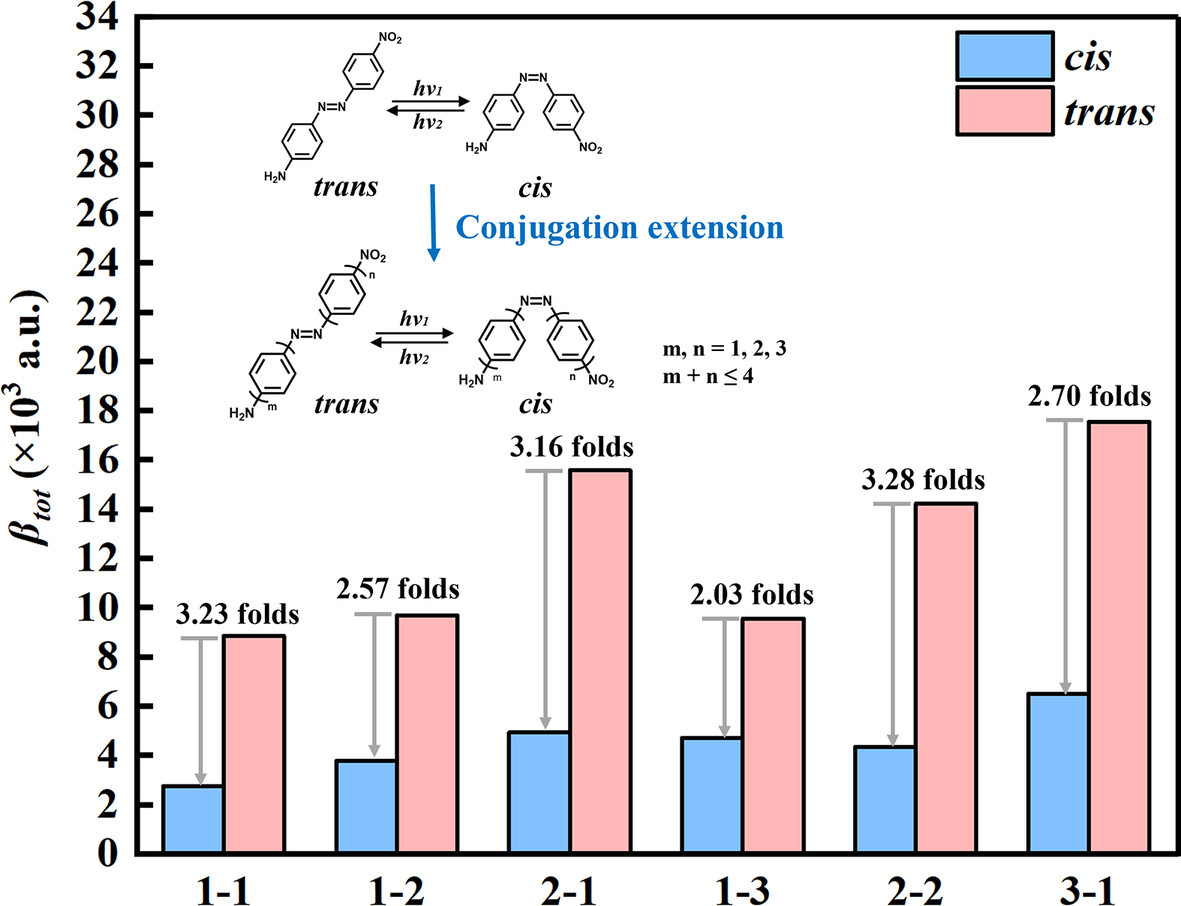
By designing a series of azobenzene derivatives with different conjugation lengths and positions of the NN bond, their nonlinear optical switching properties are verified through theoretical calculations.
Quantum Chemical, Spectroscopic and In Silico (Molecular Docking, Molecular Dynamic and ADME) Studies on Anti-Aging Pentapeptide-3 (Vialox)
- 18 July 2025
Graphical Abstract
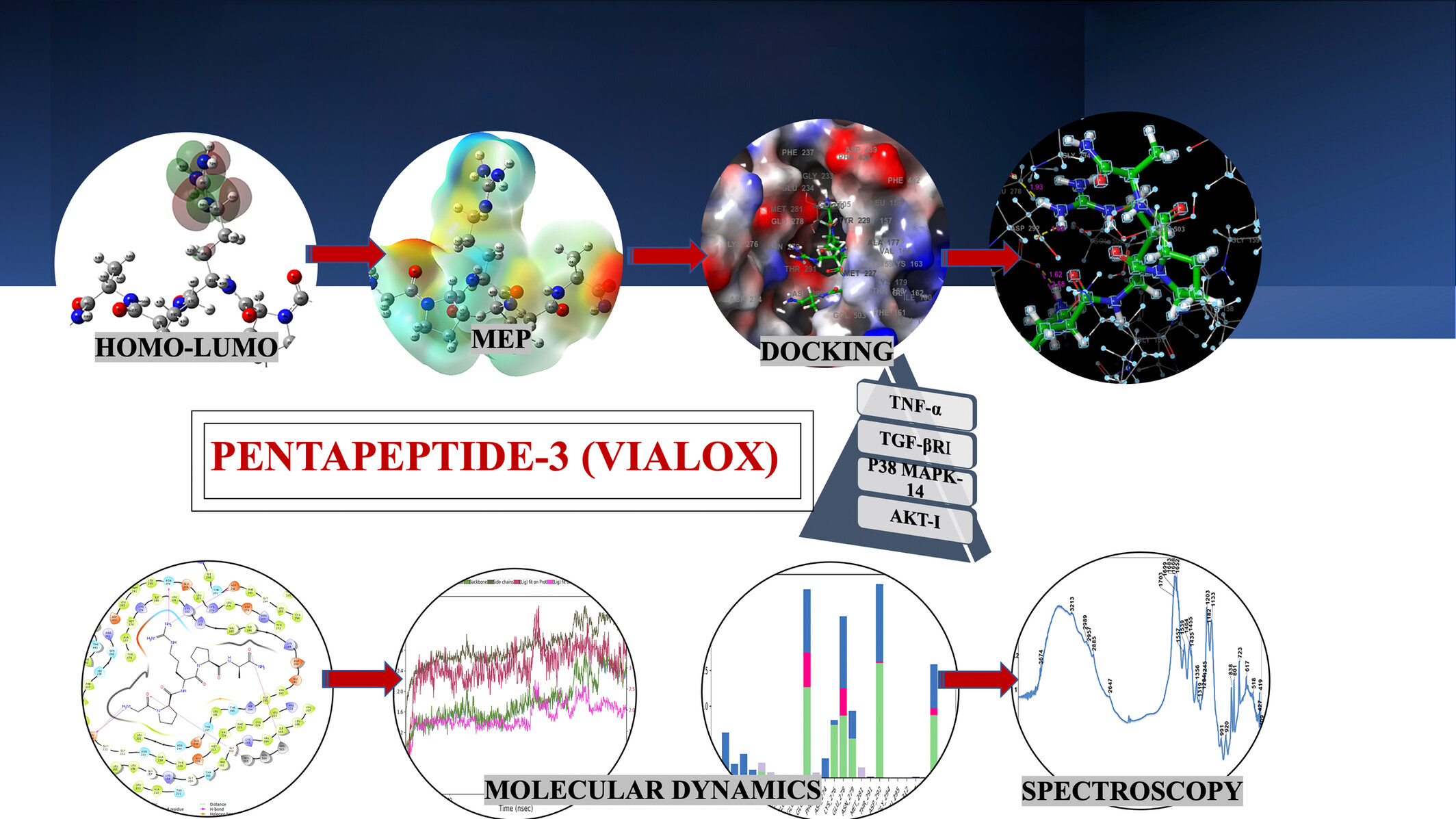
This study investigates the anti-aging potential of Pentapeptide-3 (Vialox) using quantum chemical, spectroscopic, and in silico methods. Molecular docking and dynamics analysis confirm its stable interactions with key aging-related receptors, highlighting Pentapeptide-3 as a promising candidate for cosmeceutical applications targeting skin aging.
Comparing Linear and Non-Linear Optical Properties of Symmetrical and Unsymmetrical Xanthene Dyes: DFT and TD-DFT Approach
- 14 July 2025
Graphical Abstract
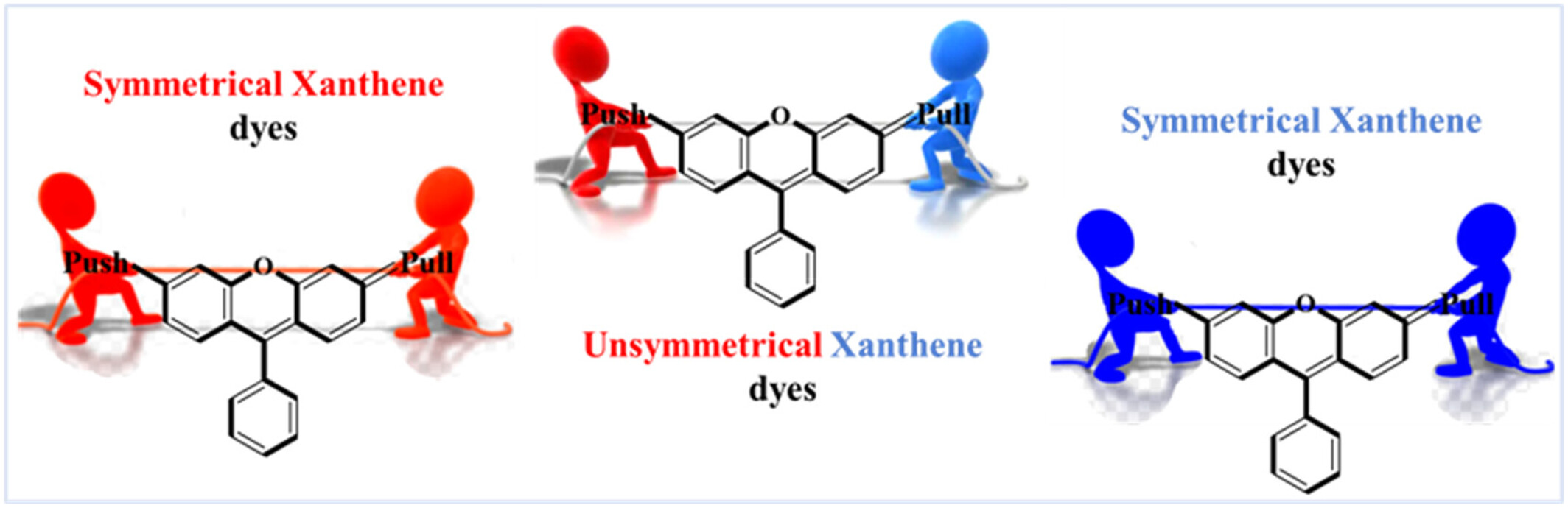
DFT results demonstrated that symmetrical xanthene dyes are energetically more stable and show relatively lower bond length alternation values. Unsymmetrical xanthene dyes with higher dipole moments and hyperpolarizability will show superior non-linear optical properties.
High Valent Mercury Hydrides
- 12 July 2025
Graphical Abstract
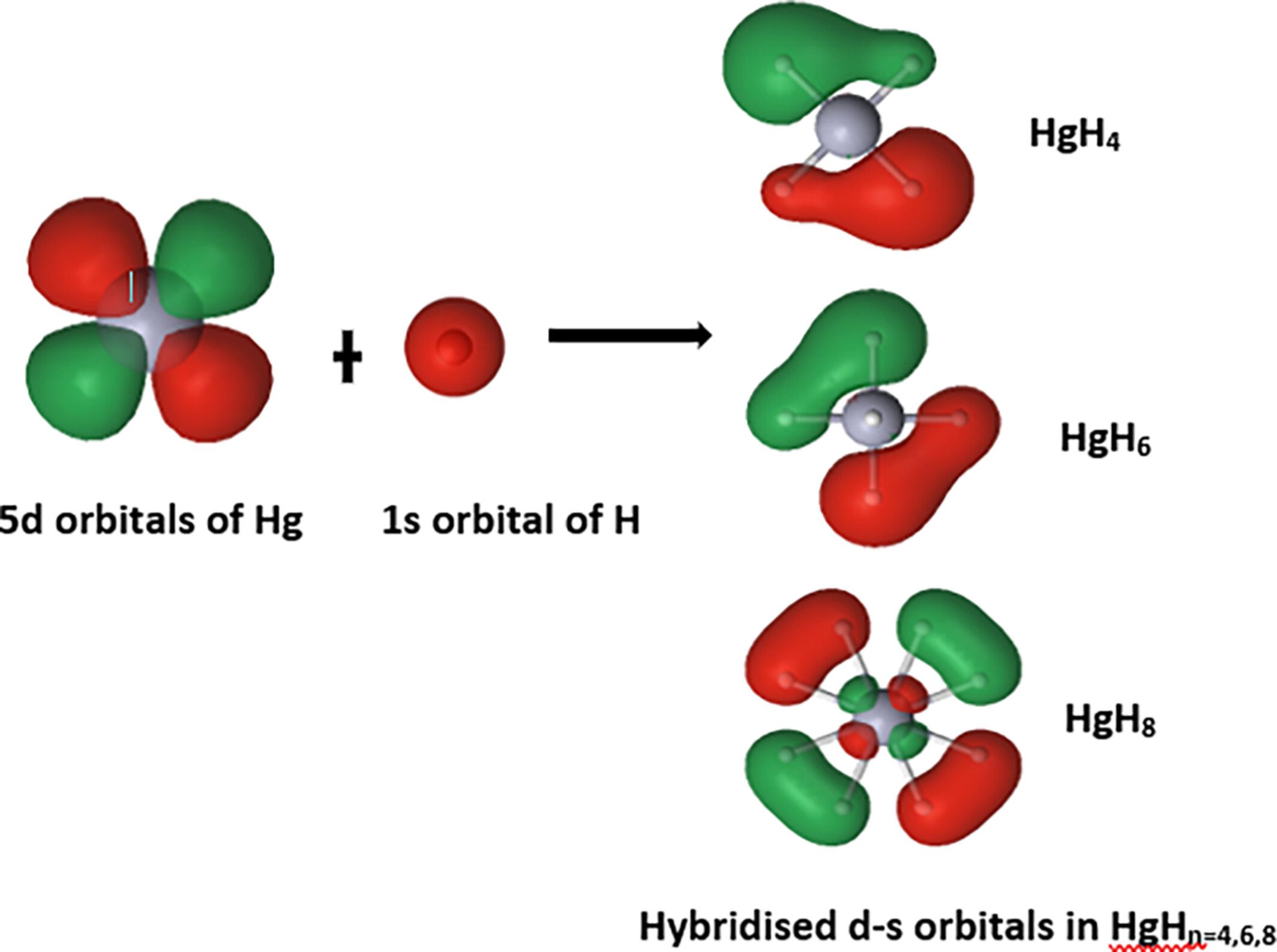
High valent mercury hydrides (HgHn) have been investigated for their structure, stability, bonding, electronic, and electrical properties using DFT-B3LYP/PBE0 and CCSD(T) methods. A large HOMO–LUMO gap (3–7 eV) higher than that of HgFn (3–5 eV) and bond capacitance of HgH (ξ(AB) = 1.52–2.09) comparable to that of CC (ξ(AB) = 1.84–2.07) bonds in benzene evidence strong covalent HgH bonds in HgHn ≥ 3. It highlights the remarkable participation of 5d Hg electrons in forming chemical bonds with hydrogen atoms.
Regulating the Photovoltaic Performance of Organic Solar Cells by Modifying the Y6-Based Non-Fullerene Acceptors: A Quantum Chemistry Study
- 12 July 2025
Graphical Abstract
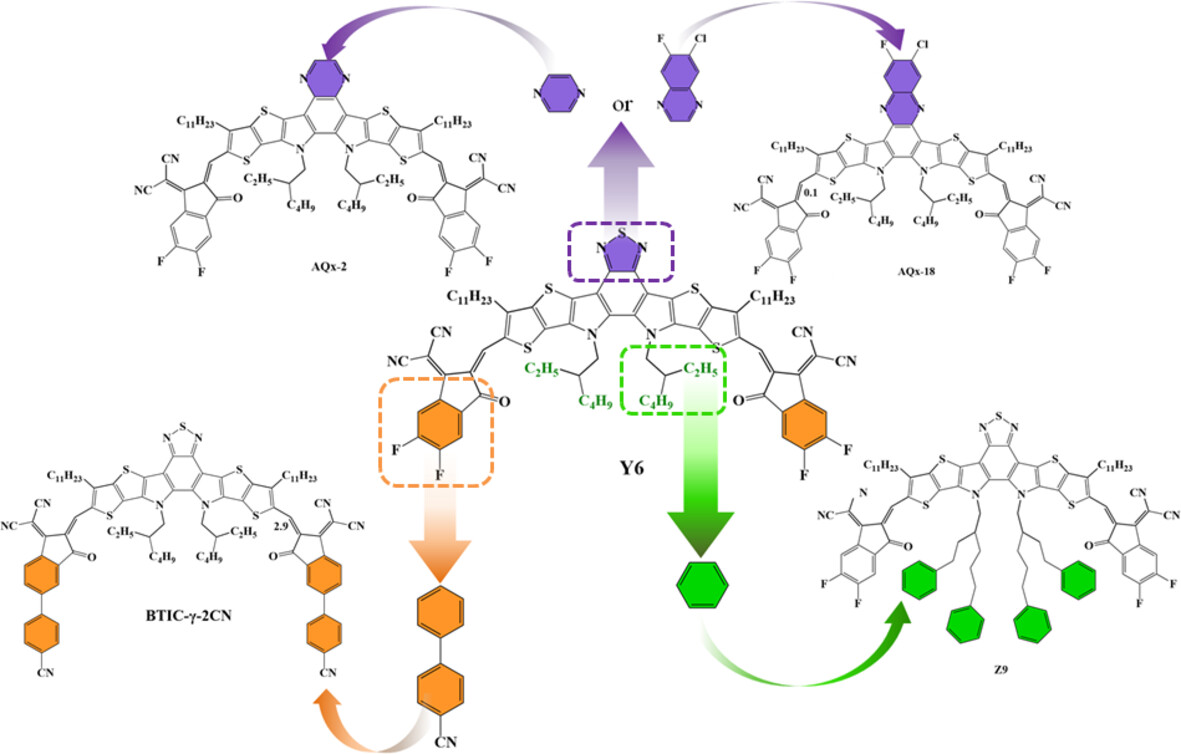
This research uses quantum chemistry calculations to study how modifying the central fused-ring, end-groups, and inner chains of Y6-based non-fullerene acceptors affects the photovoltaic performance of organic solar cells. It finds that these modifications enhance molecular planarity, adjust energy levels, and influence charge transfer and recombination processes, thus providing a theoretical basis for designing high-efficiency non-fullerene acceptors.
The following is a list of the most cited articles based on citations published in the last three years, according to CrossRef.
Jaguar: A high‐performance quantum chemistry software program with strengths in life and materials sciences
- 2110-2142
- 4 July 2013
Graphical Abstract
Jaguar is an ab initio quantum chemical program particularly suitable for applications in life and materials sciences. This work presents a comprehensive overview of the program's features and applications for the first time in its 20-year history. In addition, the review contains Jaguar timing benchmarks and discusses directions for future development of the program.
TD‐DFT benchmarks: A review
- 2019-2039
- 9 April 2013
Graphical Abstract
Time-dependent density functional theory has become the most widely used tool to investigate excited state properties. However, the selection of an adequate exchange-correlation functional remains a major issue. In this review, the results obtained through recent benchmarks are summarized and several properties considered: vertical and adiabatic transition energies, dipoles, geometries, oscillator strengths, and vibrational signatures. The review concludes with a set of general guidelines for active practitioners.
A diagnostic for determining the quality of single‐reference electron correlation methods
- 199-207
- 1/8 April 1989
Hubbard‐corrected DFT energy functionals: The LDA+U description of correlated systems
- 14-49
- 26 July 2013
Graphical Abstract
The modeling of strongly correlated materials is still a significant challenge for ab initio calculations. This article discusses the LDA+U method, reviewing its theoretical foundations, the most commonly used approximations, and recent extensions to its formulation. The aim is to highlight merits and difficulties of this approach in describing the ground state of correlated systems, and to precisely assess the conditions under which it can be expected to be most predictive.
Constructing high-dimensional neural network potentials: A tutorial review†
- 1032-1050
- 6 March 2015
Graphical Abstract
In this tutorial review, a method to construct high-dimensional interatomic potentials employing artificial neural networks is reviewed. This approach allows one to carry out molecular dynamics simulations of large systems containing thousands of atoms with close to first-principles accuracy and has been applied successfully to a number of different systems including metals, semiconductors, oxides, and molecular clusters. A strong focus of the review is on practical aspects of constructing these potentials.
Latest news
Recent issues
 Volume 125, Issue 15August 5, 2025
Volume 125, Issue 15August 5, 2025 Volume 125, Issue 14July 15, 2025
Volume 125, Issue 14July 15, 2025 Volume 125, Issue 13July 5, 2025
Volume 125, Issue 13July 5, 2025 Volume 125, Issue 12June 15, 2025
Volume 125, Issue 12June 15, 2025

