

Journal list menu

 Issue
IssueJournal of Computational Chemistry: Volume 46, Issue 20
July 30, 2025
Export Citations
Download PDFs
ISSUE INFORMATION
CORRECTION
Correction to “Three-Dimensional Metallic Boron Carbide: Stability and Properties”
- First Published: 18 July 2025
RESEARCH ARTICLE
Reduction-Induced Structural and Motional Changes of Plant-Type Ferredoxin Using Molecular Dynamics and Umbrella Sampling Simulations
- First Published: 23 July 2025
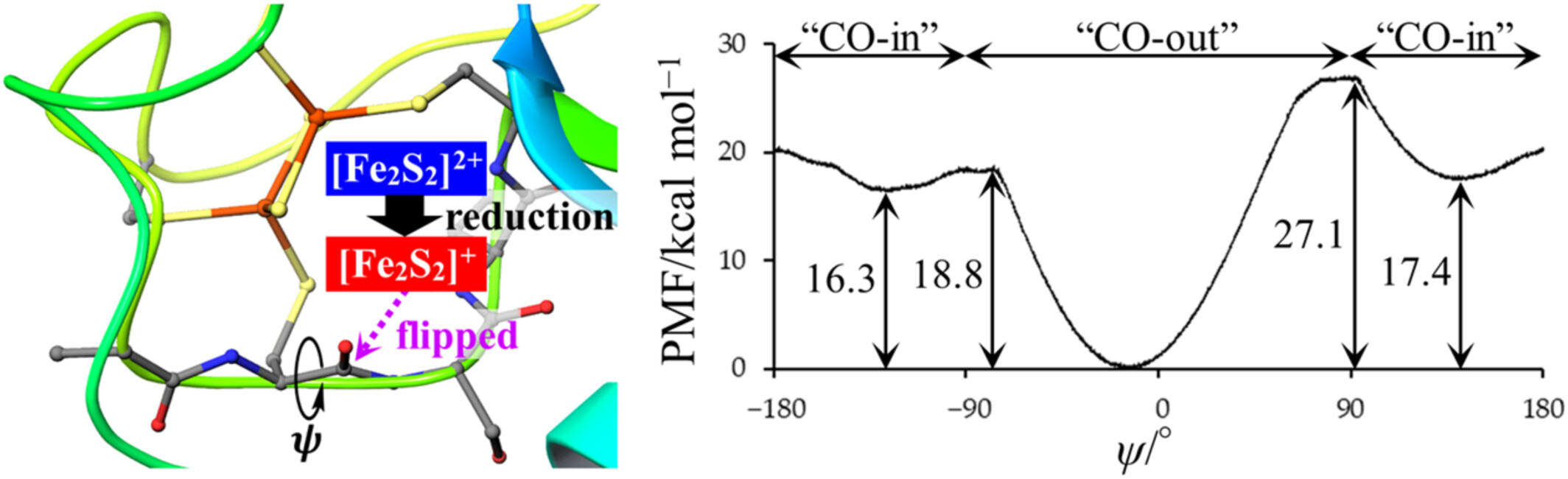
The changes of the overall structure and motion of plant-type ferredoxin associated with its active-center reduction were analyzed using long-time molecular dynamics simulations. Furthermore, the free-energy profile of peptide-bond flip induced by active-center reduction was obtained by extensive umbrella sampling simulations.
DFT Study of Structural, Chemical, and Optical Properties in Cun and PdCun−1 Clusters (n = 3–20)
- First Published: 18 July 2025
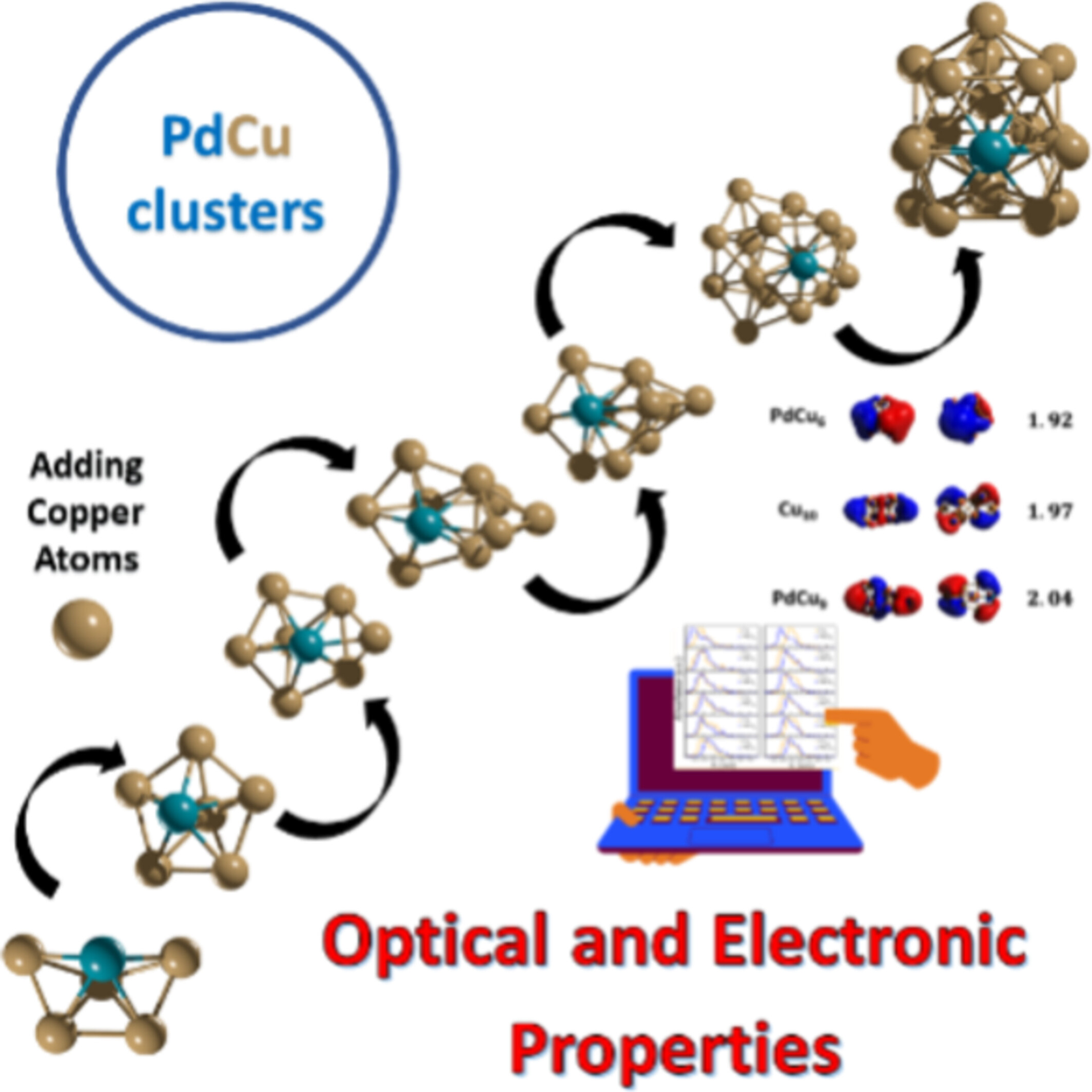
Copper and palladium–copper clusters containing up to 20 atoms are generated using a growth pattern-based algorithm. The study identifies new global minima structures and reveals how structural, electronic, chemical, and optical properties evolve with cluster size.
Topology-Induced Modifications in the Critical Behavior of the Yaldram–Khan Catalytic Reaction Model
- First Published: 18 July 2025
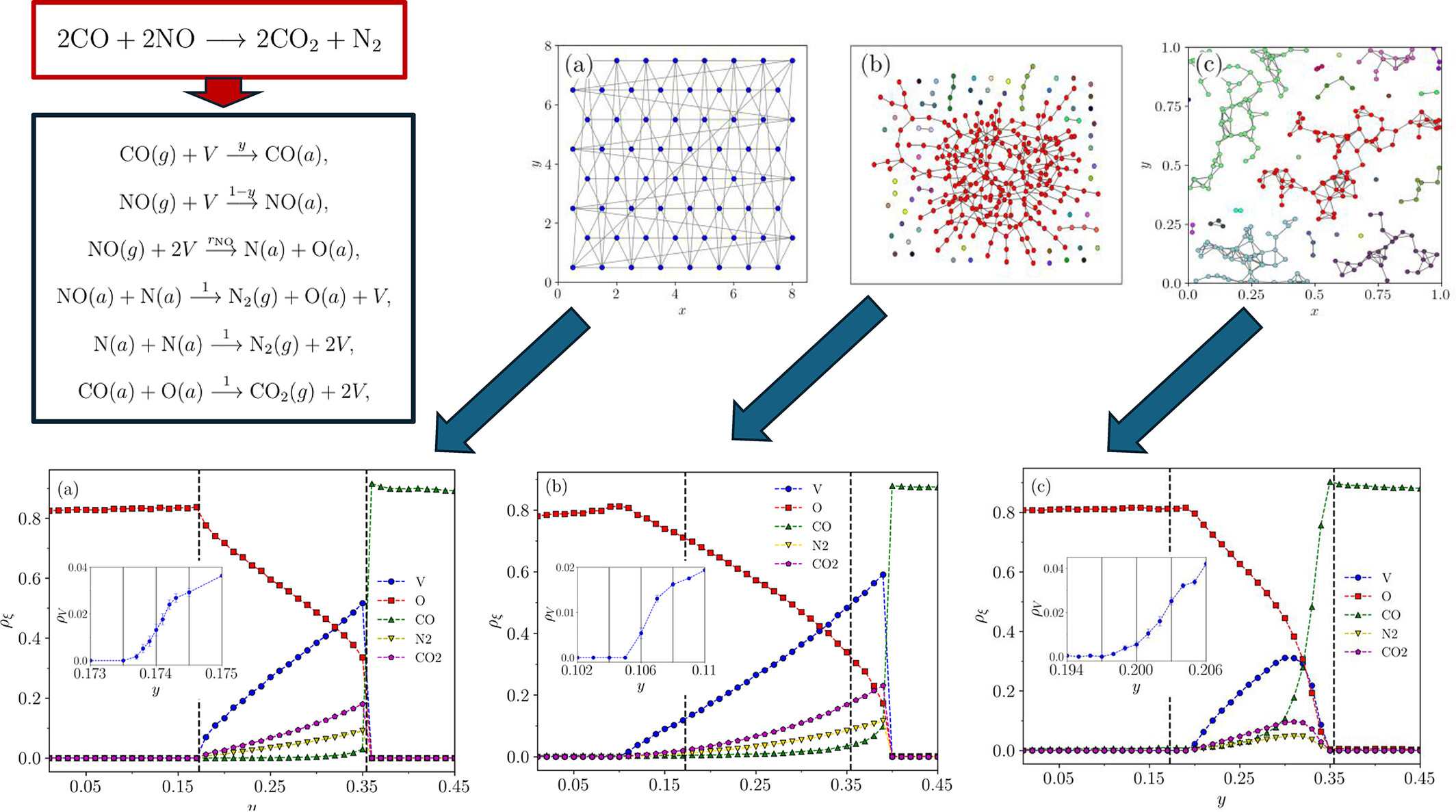
Can the phase transitions of a model be altered by changing the network topology of a catalytic reaction surface? Yes—our results demonstrate that introducing specific random-like topologies can change not only the critical points but also the order of the phase transition in the Yaldram–Khan model, suggesting promising directions for future experimental investigations.
Atomization Energy Calculations in 13-Atom Alkali Metal Clusters: Is There an Appropriate Exchange-Correlation Functional?
- First Published: 21 July 2025
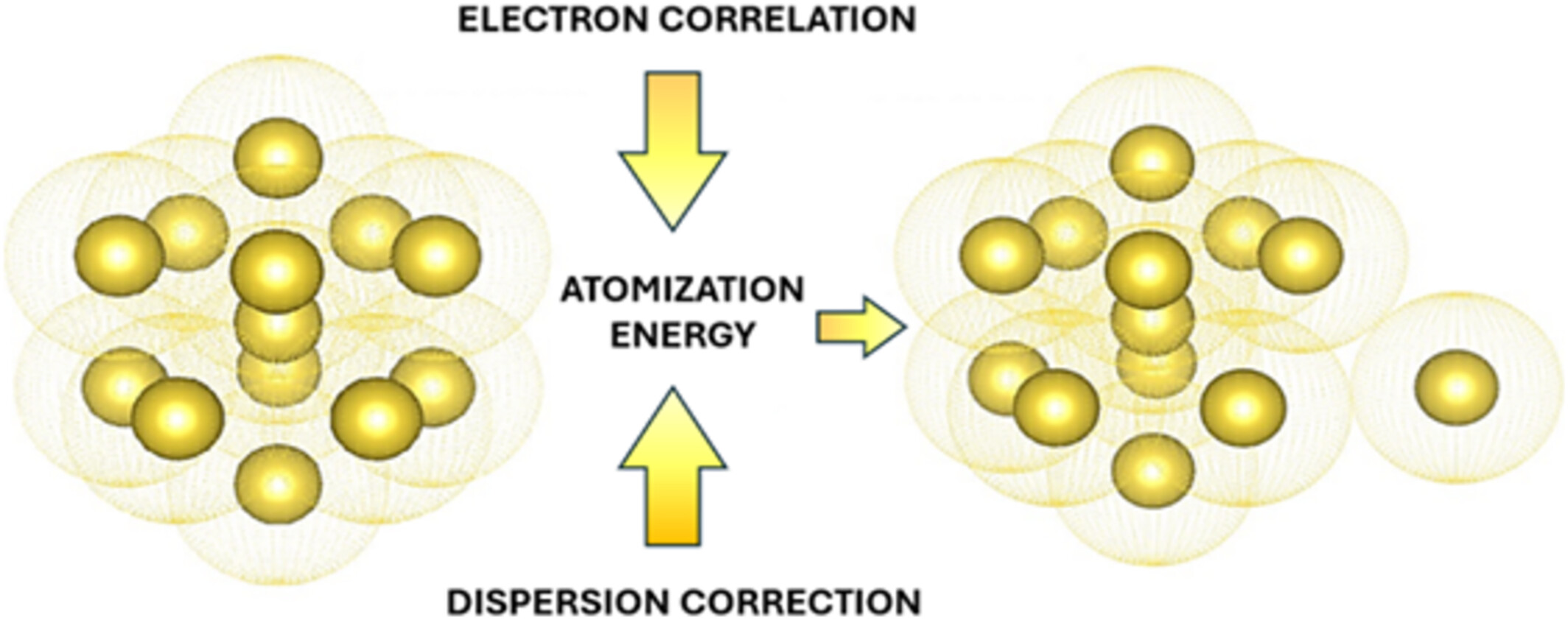
Electronic correlation and dispersion corrections are crucial for accurately modeling the atomization energies of 13-atom alkali-metal clusters within DFT.
Exact Rovibronic Equivalence of the Adiabatic and Diabatic Representations of -Coupled State Diatomic Systems
- First Published: 22 July 2025
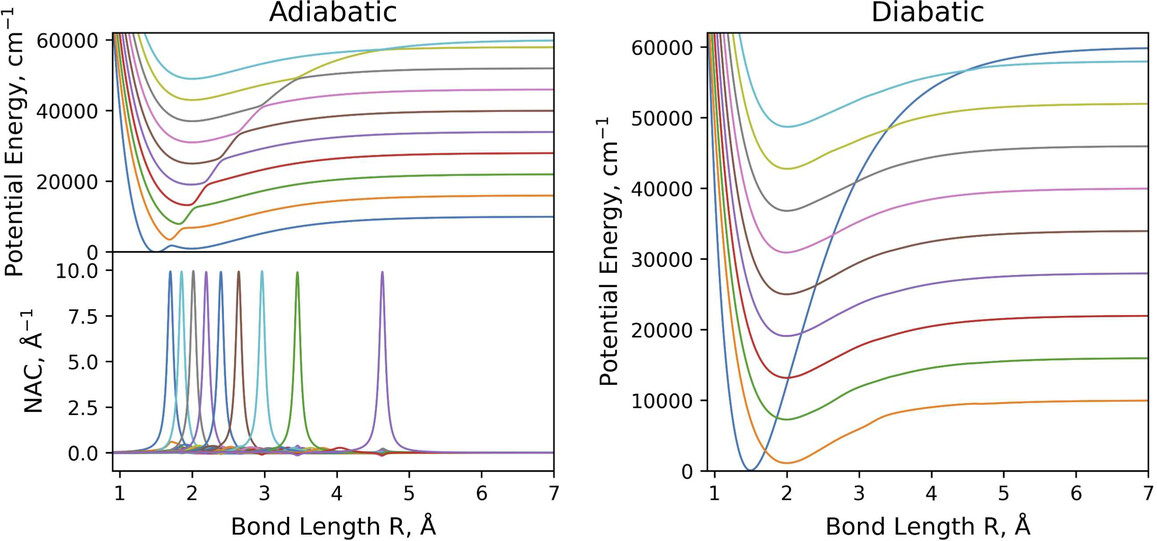
A novel method demonstrates the numerical equivalence of adiabatic and diabatic representations in N-state coupled diatomic systems. This crucial finding provides an efficient and accurate way to assess the importance of non-adiabatic effects, which are often disregarded without rigorous quantitative checks.
Ring Size Dependent Lactam-Lactim Tautomeric Equilibrium in Imidazolin-2-Chalcogenones and Pyrimidin-2-Chalcogenones: A Role of Aromaticity
- First Published: 23 July 2025
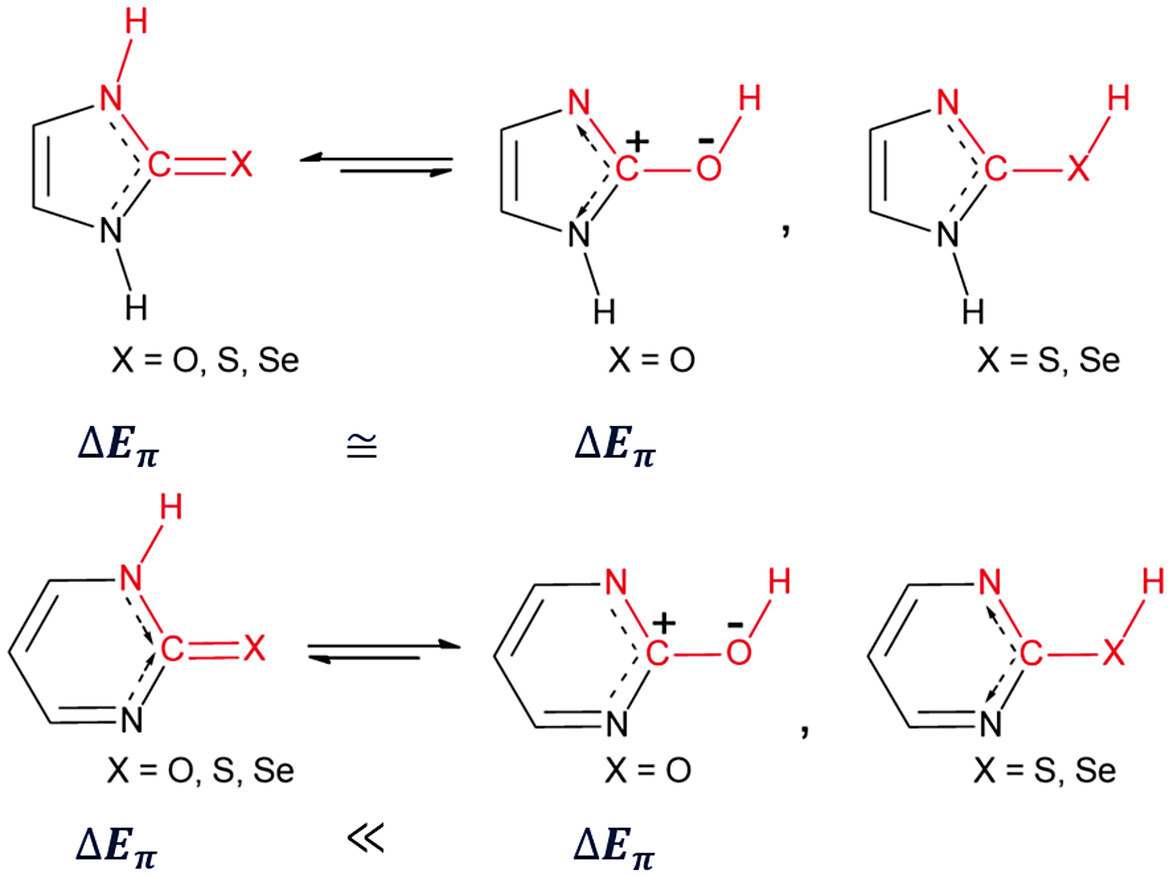
Significantly low in six-membered lactam drives the switching of the tautomeric equilibrium.
Large-Scale Calculations by Integrating the Fragmentation Approach With Neural Network Potentials
- First Published: 24 July 2025
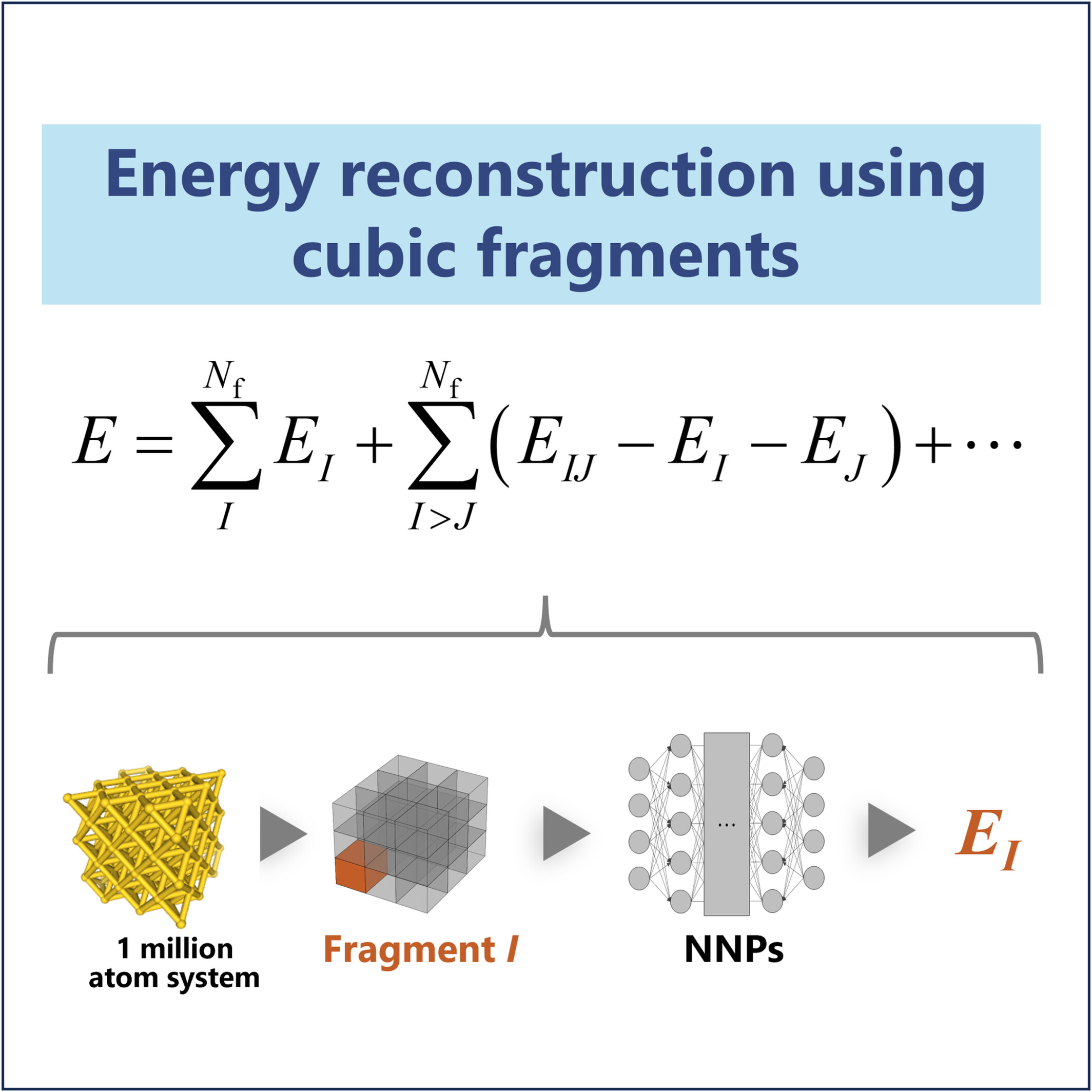
A fragmentation method is introduced to enable large-scale molecular simulations using neural network potentials. Dividing the system into fragments, the total energy is approximated using an expansion formula based on fragment interactions. An illustrative application demonstrated that the 1-million-atom systems can be handled in a reasonable computational time and with small energy errors.
Population Analysis From Variational Chemical Partition of Molecular Position Space
- First Published: 26 July 2025
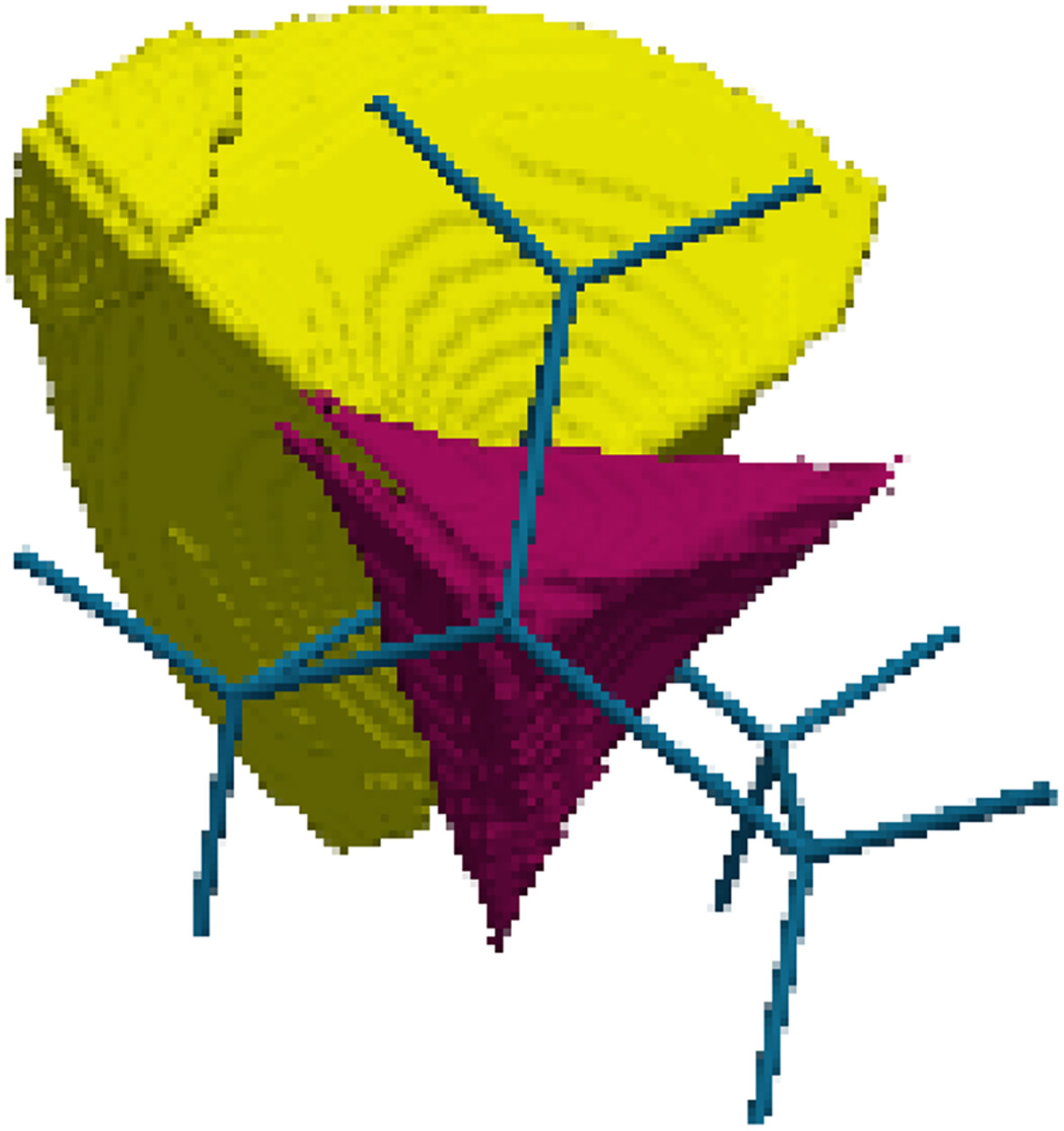
The method presented in this article divides the molecular space into electron group localization volumes within which the probability distribution of the electron numbers is underdispersed.
Two-Step Hybrid Method for the Energetics of Individual Noncovalent Interactions in Molecular Crystals
- First Published: 26 July 2025
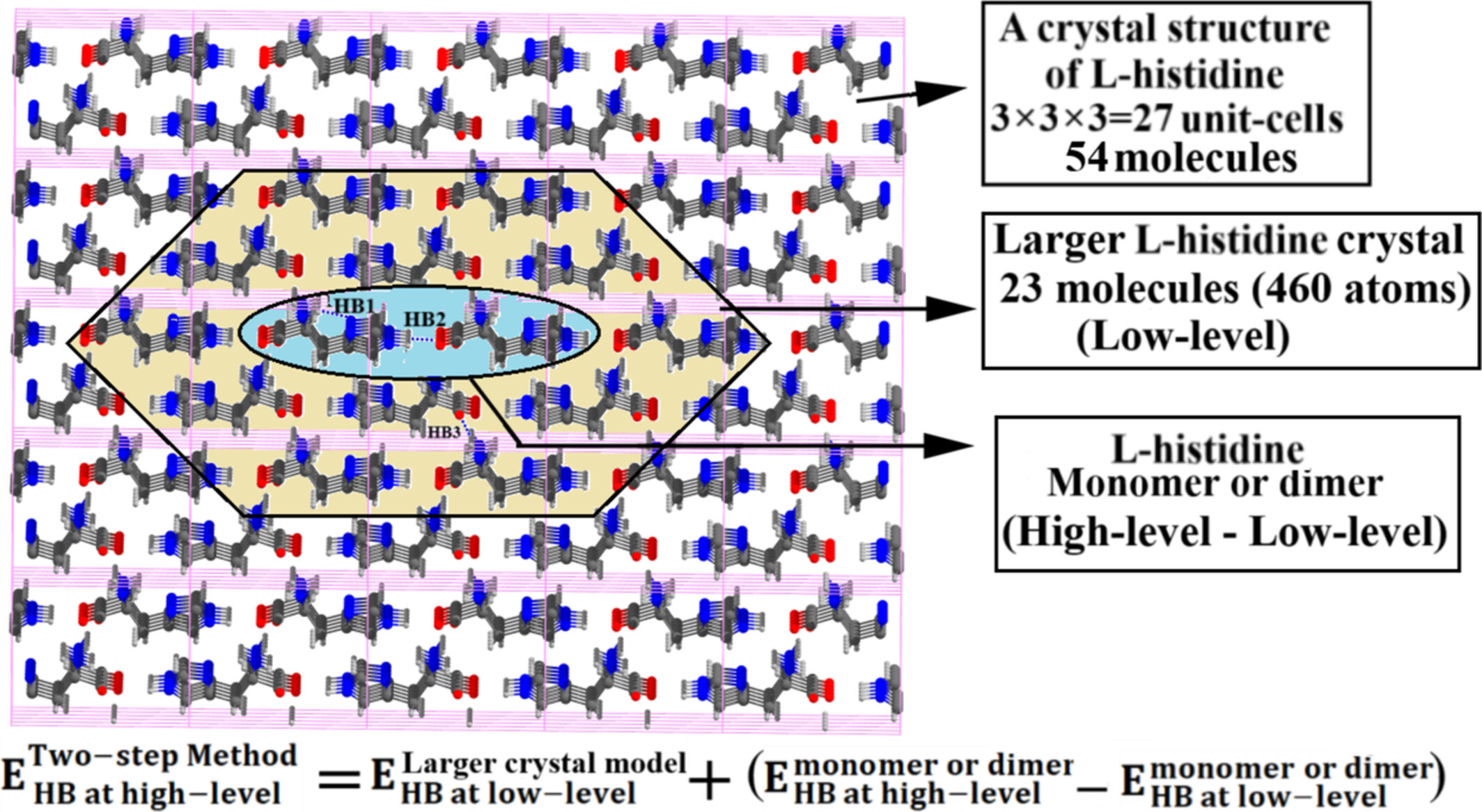
The energy of NCIs (hydrogen bond, CH…π and π…π interactions) in various molecular crystals are calculated by MTA-based and the proposed two-step hybrid methods. Application of MTA-based method is very expensive. The two-step method proposed herein, provides accurate and expeditious evaluation of energies of these NCIs in molecular crystals.
Heterogeneous Autoxidation of VOCs Promotes Light-Scattering of Atmospheric Mineral Particle: A DFT Study
- First Published: 26 July 2025
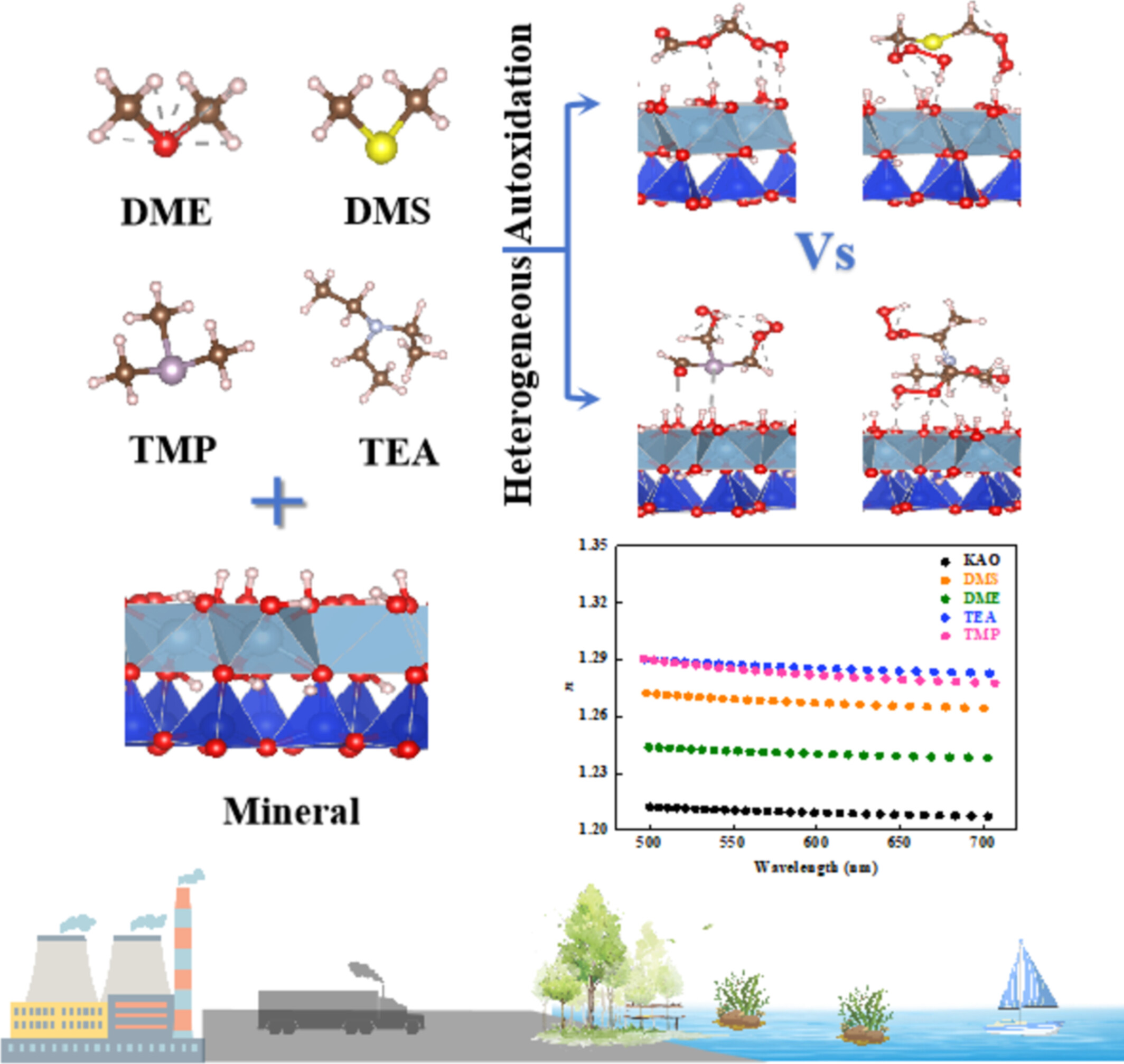
Heterogeneous autoxidation of volatile organic compounds (VOCs) significantly enhances the light-scattering efficiency of mineral particles, with alkane-type VOCs exhibiting superior enhancement relative to ether-type analogs.
Sign up for email alerts
Tools
