

High Valent Mercury Hydrides
Funding: The author received no specific funding for this work.
ABSTRACT
High valent mercury hydrides (HgHn=1–80,±) are studied using DFT-B3LYP/PBE0 and CCSD(T) methods to analyze their structure, stability, bonding, electronic properties, and electrical characteristics. Molecular orbital calculations reveal a high HOMO–LUMO energy gap of ≈ 7 eV for HgH2, HgH4, and HgH6, while HgH8 shows a lower energy gap of ≈3 eV. The HgH bond capacitance (ξ(AB)) values are 1.84, 2.03, 2.09, and 1.52 for HgH2, HgH4, HgH6, and HgH8, at the PBE0 level. These values are comparable to the CC bond capacitance range (ξ(AB) = 1.84–2.07), providing evidence for stable high-valent HgHn with oxidation states ranging from I to VIII. Furthermore, they show a linear response to an external static electric field up to 0.1 a.u., with a gradual decline for higher fields. These observations indicate their possible future detection through molecular beam deflection or the matter-wave interferometry technique.
1 Introduction
The relativistic mass of the electron in the Dirac-Hamiltonian perturbs the orbital energy levels of atoms through mass–velocity contraction, spin-orbit coupling, and Darwin's contract term [1-10]. Consequently, the 6s and 5d atomic orbitals in the mercury (Hg) atom undergo contraction and expansion respectively [11]. Various theoretical methods predicted the participation of 5d10 shell electrons in chemical bonding with highly electronegative atoms, such as fluorine (F) [12-17]. The involvement of 5d electrons in chemical bonding was recently confirmed in HgFn0/± through vibrational spectroscopy [18] and mass spectrometry [19].
The interaction between the Hg and the hydrogen (H) atom offers a unique opportunity to explore the effects of the special theory of relativity on Hg through “sd” hybridization. Quantum chemical calculations predicted the possibility of HgH4 and HgH6 with Hg in +IV and VI oxidation states (OS) [20]. However, other molecules in the HgHn series like HgH3, HgH5, HgH7, and HgH8 and their corresponding ions remain unexplored. We have systematically investigated HgHn=1–80± for bonding, stability, electronic properties, and electrical characteristics by quantum chemical methods to support their possible detection through molecular beam deflection or matter-wave interferometry technique.
2 Computational Methods
The molecular structure of HgHn=1–80± is optimized using the density functional theory (DFT) based method at B3LYP/PBE0 hybrid electron exchange-correlation functional [21-24] and wave functional theory-based coupled cluster-single-double and perturbative triple [CCSD(T)] level [25]. Atomic natural orbital with relativistic core corrections and quadrupole-ζ plus polarization (ANO-RCC-VQZP) basis set with the following primitive/contracted sets: 25s22p16d12f4g2h/9s8p6d4f2g1h and 14s9p4d3f2g/5s4p3d2f1g is used for Hg [26] and H [27]. The scalar relativistic effects are included through one-electron integrals in ANO basis sets using the X2C (exact two-component) method [28-31]. The spin-orbit coupling (SOC) corrections are not calculated due to their negligible contribution to similar systems, HgXn (X = F, Cl, Br, I) [32], although SO splitting of 5d orbitals in Hg is 2.052 eV at the Dirac-Fock level [4]. The resolution identity (RI) decomposed two-electron repulsion integrals are stored in Cholesky decomposition (CD) style to reduce the computation time [33, 34]. The vibrational frequency analysis is also carried out within harmonic approximation to identify the nature of minima on the potential energy surface (PES). The stability of HgHn is determined using the atomization energy, transition state, and the energy gap between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO). The isotropic polarizability and bond capacitance are computed for a range of electric field strengths using the LoProp method. All the calculations are performed using MOLCAS 8.0 [35] and CFOUR 2.1 software [36].
3 Results and Discussion
3.1 Structure of HgHn
A stable HgH2/HgD2 has also been prepared by the reaction between excited Hg and H2 gas in H2/D2, H2/Ar, and H2/N2 matrices. The product molecules formed are detected and characterized using IR spectroscopy supported by quantum chemical calculations. Moreover, by annealing processes, (HgH2)2, (HgH2)3, and solid HgH2 were observed in solid hydrogen in a low-temperature matrix isolation technique [40-43]. The stability of (HgH2)n is attributed to the closed shell Hg2+–Hg2+ mercurophilic interaction that arises from the relativistic effects and other noncovalent dispersive forces [44, 45]. Additionally, gaseous HgH2/HgD2 is prepared through the direct gas phase reaction of Hg vapor with H2/D2 in an electrical discharge. High-resolution infrared emission spectra reveal a linear covalent HHgH molecule with a HgH bond length of 1.646543 Å and Hg in a typical +II oxidation state [46, 47]. Theoretical calculations predicted a bond length of 1.615–1.713 Å with real frequency values [48-55]. In the past, HgH4 and HgH6 have been investigated computationally for high OS by employing DFT-B3LYP, MP2, and CCSD(T) methods [20]. However, structural and spectroscopic data for other members in the HgHn family and their corresponding ions have not yet been reported. Table 1 lists the computed structural data, symmetry, IR frequency, ionization potential (IP), and electron affinity (EA) of HgHn=1–80,± at the DFT-B3LYP/PBE0 and CCSD(T) levels.
| HgHn=1–80/± | Structure | Point group | HgH bond lengths (Å) B3LYP/PBE0/{CCSD(T)} | HHgH bond angle (degree) B3LYP/PBE0/{CCSD(T)} | Harmonic vibrational frequency (cm−1) B3LYP/PBE0/{CCSD(T)} | Ionization energy (eV) B3LYP/PBE0/CCSD(T) | Electron affinity (eV) B3LYP/PBE0/CCSD(T) |
|---|---|---|---|---|---|---|---|
| HgH |  |
C∞v | 1.776/1.742/1.686 | 1041/1346/{1606} | 8.164/8.022/7.978 | 1.093/0.904/0.873 | |
| HgH+ |  |
C∞v |
1.607 (1.610)a/1.600/{1.560} |
1753 (1987)a/1775/{2178} (2028)b | |||
| HgH− |  |
C∞v | 2.107/1.981/{1.890} | 788/780/{1049} | |||
| HgH2 |  |
D∞h |
1.654 (1.653)a/1.647/{1.604} |
180.00/180.00/{179.99} |
1625, 1964, 2070/826, 1954, 2073/{872, 872, 2099, 2246} |
10.589/10.533/10.795 | −0.448/−0.491/−0.603 |
| HgH2+ |  |
C2V | 1.713/1.704/{1.667} | 148.23/145.92/{179.99} | 2 89,1485, 1863/3 12,1 453, 1868/{xxx} | ||
| HgH2−, |  |
D∞h | 1.658/1.652/{1.609} | 180.00/180.00/{179.99} |
4 15,1910, 2055/4 73,1908, 2058 {529,529,2028, 2205} |
||
| HgH3 |  |
C2V | 1.657, 1.886/1.650, 1.868/{1.671} | 92.66,174.67/92.59,174.82/{119.99} | 7 41,854, 927,942,1969, 2046/740,863,9 03,918,1963, 2051/{xxx} | 6.243/6.213/10.512 | 2.226/2.357/3.250 |
| HgH3+ |  |
C2V | 1.593, 2.075/1.587, 2.025/{1.734} | 21.51169.25/22.28168.86/{119.99} |
532.539,683,8 64,1944, 3967/539, 569, 6 96,898,2007, 3897 /xxxx |
||
| HgH3− |  |
D3h | 1.764/1.752/{1.702} | 120.00/120.00/{119.99} |
i1465, 332, 637, 1400, 1449, 1540/i2564, 343,646,1478,1496, 1620 {342,342,640,1692, 1692, 1802} |
||
| HgH4 |  |
D4h |
1.637(1.641)a/1.628/{1.592} (1.635)a |
90.00/90.00/{90.00} |
503,737,737,8 65,908,1947, 1947, 2089, 2107/677,738,7 39,877,927,1971, 1971, 2089, 2114 {869,869,874,892,921,2142, 2142, 2262, 2262} {764,759,839,884,1959, 2060, 2085}a |
9.870/9.681/11.341 | −0.192/−0.236/−0.236 |
| HgH4+ |  |
D2h |
1.707/1.701/{1.627} |
40.56,139.54/40.03,139.97/{90.00} |
i1503, 140, 580, 819, 843,1450, 1754, 1886, 1957 /i1420, 158, 578, 861, 928, 1520, 1793, 1934, 2008 {xxx} |
||
| HgH4− |  |
D4h | 1.637/1.628/{1.592} | 90.00/90.00/{90.00} |
i4348, 616,718,747,792,1823, 1893, 2010, 2043 /i2144, 668,710,743,769,1913, 1915, 2045, 2075/{xxx} |
||
| HgH5 |  |
C4V | 1.638, 2.149/1.629, 2.371/{1.616} | 89.98, 90.97/90.00, 90.52/{90.00} |
i301, i263, 332, 666, 726, 735, 885, 893, 1934, 1939, 2054, 2123/143, 443, 445, 714, 723, 726, 862, 900, 1969, 1970, 1993, 2097 {xxx} |
6.474/10.207/9.974 | 2.381/2.246/2.094 |
| HgH5+ |  |
C2V | 1.563, 1.654, 1.967/1.647, 1.541/{1.594} |
23.38, 82.38, 85.93/87.30, 89.97/{90.00} |
426, 457, 495, 606, 800, 854,907,1148, 1973, 2010, 2183, 3630 /i1048, i1043, 718, 735, 753, 928, 929,1952, 1990, 2013, 2031, 2191/{xxx} |
||
| HgH5− |  |
C4V | 1.655, 1.908/1.645, 1.885/{1.632} | 88.81, 98.28/88.74, 98.54/{90.00} |
26, 586, 625, 626, 671, 686, 689, 1217, 1751, 1810, 1874, 1949/120, 566, 611, 644, 662, 735, 737, 1267, 1760, 1838, 1951, 1989 /{xxx} |
||
| HgH6 |  |
Oh |
1.632(1.636)a/1.622/{1.592} (1.635)a |
90.00/90.00/{90.00} |
i2522, i1742, 659, 697, 720, 803, 804, 808, 1909, 1941, 1943, 2032, 2065, 2354, 3126 /i3979, i1009, 691,696,698,810,813,823,1923, 1964, 1984, 1991, 2075, 2195, 4425 {757,757,757,830,830,830,2051, 2051, 2051, 2238, 2283, 2283} {695, 755, 810, 1984, 2063, 2090}a |
10.273/10.154/11.694 | 0.550/0.541/−0.273 |
| HgH6+ |  |
D3d | 1.665/1.655/{1.622} | 70.51/69.95/{90.00} |
i8574, i5921, i322, 360, 387, 608, 609, 667, 723, 1745, 1748, 1777, 1908, 6014, 8571 /i9026, i4127, i423, 320, 353, 598, 606, 671, 731, 1485, 1767, 1775, 1818, 2708, 8222/{xxx} |
||
| HgH6− |  |
D4h | 1.629, 1.787/1.637, 1.958/{1.667} | 90.00/90.00/{90.00} | i1628, i1541, 513, 527, 527, 573, 720, 720, 7 507 501 395, 1397, 1490, 1849, 2042/435, 435, 574, 637, 664, 664, 677, 686, 686, 952, 1046, 1866, 1880, 1880, 1975/{xxx} | ||
| HgH7 |  |
C2V | 1.638, 2.195, 3.613/1.629, 2.417, 3.236/{1.715} | 11.79, 85.01, 90.00, 90.83/13.23, 84.93, 90.04, 90.40/{45.00} | 58, 98, 105, 151, 267, 281, 292, 296, 706 714, 727, 798, 854,1940, 1940,2042, 4412, 11358/92, 94, 98, 148, 179, 206, 300, 308, 725, 726 760, 858, 875,1960, 1961, 2079, 4387, 11049/{xxx} | 9.301/9.109/NC | 2.375/2.226/2.353 |
| HgH7+ |  |
C2 | 1.552, 1.650, 1.654, 1.952/1.547, 1.641, 1.643, 1.909/{NC} |
23.64, 86.61, 86.96, 91.65/24.62, 86.01, 86.80, 90.24/{NC} |
i3868, i263, i190, 389, 622, 749, 778, 840, 860, 900, 1158, 1323, 1936, 1963, 2015, 2229, 3553, 5593 /i166, i107, 163, 486, 515, 747, 756, 851, 860, 968, 1192, 1292, 1947, 1969, 1974, 2272, 3472, 5151/{NC} |
||
| HgH7− |  |
C2V | 1.656, 1.908, 4.542/1.645, 1.884, 4.307/{1.786} | 9.36, 78.31, 88.78, 98.39/9.91, 77.91, 84.91, 98.61/{44.99} |
i1237, i239, i180, 48, 59, 81, 542, 587, 594, 668, 674, 679, 1215, 1752, 1754, 1868, 4421, 7587/ i1030, i262, i127, i106, 58, 105, 551, 566, 583, 663, 667, 672, 1266, 1758, 1759, 1895, 4405, 7730/{xxx} |
||
| HgH8 | 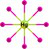 |
D8h | 1.997/1.982/{1.795} | 45.00/45.00/{44.99} |
i2256, i2256, i1437, i1436, i1273, i637, i634, i532, i530, i245, 623, 623, 966, 968, 1034, 1039, 1039, 1195, 1195, 1319, 1620 /i2286, i2285, i1410, i1409, i1209, i632, i633, i571, i570, i248, 712, 713, 978, 985, 1057, 1083, 1083, 1248, 1257, 1367, 1679/{xxx} |
−2.944/−2.466/xxx | xx/xx/3.099 |
| HgH8+ | 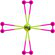 |
D2h | 2.701/2.652/{1.743} | 15.99, 74.01/16.37, 73.62/{44.99} |
i1371, i443, i120, i108, i106, i60, i19, 43, 81, 125, 192, 239, 289, 293, 323, 703, 1124, 4252, 4258, 4258, 4278 /i3354, i468, i115, i109, i55, i45, i15, 58, 100, 124, 229, 247, 295, 317, 346, 600, 3331, 4234, 4234, 4250, 4252//{xxx} |
||
| HgH8− | 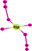 |
Cs | NC/NC/{1.843} | NC/NC/{44.99} | NC/NC/{xxx} |
3.1.1 HgH
HgH is a linear molecule with C∞v symmetry and shows an equilibrium bond length (re) of 1.776/1.742/1.686 Å at B3LYP/PBE0/CCSD(T) levels. The re value calculated at the B3LYP level agrees with the experimental value (1.776 Å) [37] and earlier reported theoretical values (1.790, 1.796 Å) at the same level of theory [38, 44]. The HgH+ ion shows shortened re of 1.607/1.600/1.560 Å and agrees with the experimental value (1.594 Å) [42] and also the reported theoretical values (1.610/1.579/1.607 Å) [20] at the B3LYP/MP2/CCSD(T) levels with an IP of 8.164/8.022/7.978 eV. While its negative ion (HgH−) shows elongated re of 2.107/1.981/1.890 Å with a positive EA value of 1.093/0.904/0.873 eV, indicating that the anion is more stable than neutral. The molecule shows a frequency of 1041/1346/1606 cm−1 with intensity of 94/113/107 km mol−1. The computed zero-point vibrational energy (ZPVE) is 520/673/803 cm−1. The molecule and the ions show real frequency values, indicating they are stable species.
3.1.2 HgH2
HgH2 is a linear molecule with D∞h symmetry and shows re of 1.654/1.647/1.604 Å at B3LYP/PBE0/CCSD(T) levels and is in agreement with the earlier reported values at different levels of theory viz., 1.656/1.617/1.641 Å at B3LYP/MP2/(CCSD(T)) levels [20, 42-46]. The cation shows a bend geometry with C2V symmetry and slightly elongated re of 1.713/1.704/1.667 Å and ∠HHgH = 148.23/145.920 at B3LYP/PBE0 level while (CCSD(T)) shows linear structure with an angle of 180°. The anion also shows a linear structure with re of 1.658/1.652/1.609 Å. The linear HgH2 shows 3N-5° of freedom and shows 4 normal modes of vibration with A1, B1, B2, and A1 irreducible notation. All the modes show real frequency and are IR active except 1A1. Among the three IR active modes, B2 shows a high IR frequency of 1964/1954/2099 cm−1 with an intensity of 434/411/373 km mol−1 and ZPVE of 2829/2427/2608 cm−1. The HgH20/± systems show real frequency values indicating they are stable on the PES. The molecules show an IP of 10.589/10.533/10.795 eV and an EA of −0.448/−0.491/−0.603 eV. The negative EA value indicates that the anion is less stable than the neutral molecule.
3.1.3 HgH3
HgH3 exhibits T-shaped trigonal planar geometry with C2v symmetry and shows two distinct types of bonds with re of 1.657, 1.886/1.650, 1.868 Å along with ∠H1HgH2 = 174.67/174.82° and ∠H2HgH3 = 92.66/92.59° at the B3LYP/PBE0 level. In contrast, the CCSD(T) method shows an equal bond length of 1.671 Å with ∠HHgH = 120°. The cation shows a short HgH of re 1.593/1.587 Å and a long HgH of re of, 2.075/2.025 Å along with reduced bond angles at B3LYP/PBE0 level and it exists as HgH+ and H2. The CCSD(T) method shows elongated re of 1.734 Å and ∠HHgH = 120° and it exists as HgH3+. The anion shows slightly elongated re of 1.764/1.752/1.702 Å and ∠HHgH = 120° at all levels of theory. The degrees of freedom for non-linear HgH3 are calculated using the formula 3N-6. Accordingly, HgH3 shows 6 normal vibration modes with A′1, A′2, E′, A″1, A″2, E″ irreducible representation in the point group symmetry with IR active E′ and A″2 modes. All the vibrational modes show a real frequency. The IR frequency for an active mode A″2 is 1969/1963 cm−1 with an intensity of 407/391 km mol−1. The ZPVE is computed to be 3740/3720 cm−1. The positive ion shows real frequency while the negative ion shows one imaginary frequency, indicating a stable cation and unstable anion. Both B3LYP/PBE0 levels show a low IP of 6.243/6.213 eV and EA of 2.226/2.357 eV while CCSD(T) calculations show a high IP of 10.512 eV and high EA of 3.250 eV. The relatively high EA suggests that HgH3 is stabilized by electron capture and supports the existence of OS III in HgH3 (5d9), however, it is vibrationally unstable except at the CCSD(T) level.
3.1.4 HgH4
HgH4 is a square planar molecule with D4h symmetry and shows re of 1.637/1.628/1.592 Å at B3LYP/PBE0/CCSD(T) levels along with ∠HHgH = 90°. The re values agree with the reported values, 1.641/1.618/1.635 Å, and ∠HHgH = 90° at B3LYP/MP2/CCSD(T) levels [20]. The cation displays an elongated re of 1.707/1.701 Å and shows a decrease in bond angles and the formation of 2H2 molecules at the B3LYP/PBE0 level, while CCSD(T) shows intact HgH4+ with re of 1.627 Å. The anion shows the same bond lengths and bond angles as the neutral molecule. HgH4 shows 9 normal vibration modes with 2Eu, 1A2u, 1B1g, 1B1u, 2Eu, 1A1g, 1B2g irreducible notation in the point group symmetry. All the normal modes show real frequency but are IR inactive except the Eu modes. The IR active Eu modes show a frequency of 2089/2089/2142 cm−1 with an intensity of 354/337/324 km mol−1. The computed ZPVE is 5921/6051/6634 cm−1, evidencing vibrationally stable HgH4 with Hg4+(5d8) OS while its ions show one imaginary frequency, indicating they are not a stable minimum on the PES. An IP of 9.870/9.681/11.341 eV and an EA of −0.192/−0.236/−0.236 eV is calculated for HgH4. The negative EA also indicates that the anion is less stable than the neutral molecule.
3.1.5 HgH5
HgH5 exhibits square pyramidal geometry with C4v symmetry and displays two different types of bonds with re of 1.638, 2.149/1.629, 2.371 Å along with ∠H1HgH2 = 90.97/90.52°, ∠H3HgH4 = 89.98/90.00°, and ∠H5HgH6 = 89.98/90.00° at the B3YLP/PBE0 level. It exists as an HgH4…H complex with a large re of 2.149/2.371 Å rather than an HgH5 molecule. In contrast, the CCSD(T) shows an equal re of 1.616 Å for all bonds with ∠HHgH = 90° suggesting HgH5 exists as a molecule. The cation shows three unequal HgH bonds with re of 1.563, 1.654, 1.967 Å at the B3LYP level and PBE0 shows two unequal bonds with re of 1.647, 1.541 while CCSD(T) shows re of 1.594 Å. The anion shows two unequal bonds with re of 1.655, 1.908/1.645, 1.885 Å at B3LYP/PBE0 levels while CCSD(T) shows re of 1.594 Å. The IR frequencies for the vibrational modes computed at the B3LYP level show one imaginary frequency while PBE0 shows real frequency. The positive ion shows the real frequency at the B3LYP level and an imaginary frequency at the PBE0 level. The negative ion shows real frequency at both levels of theory. An IP of 6.474/10.207/9.974 eV and an EA of 2.381/2.246/2.094 eV have been noted for HgH5. The positive EA value indicates that the HgH4…H complex is further stabilized through electron capture processes.
3.1.6 HgH6
HgH6 shows octahedral geometry with Oh symmetry and re of 1.632/1.622/1.592 Å along with ∠HHgH = 90° at the B3LYP/PBE0/CCSD(T) levels, which is in agreement with the reported re values of 1.636/1.619/1.635 Å at B3LYP/MP2/CCSD(T) levels [20]. The cation shows slightly elongated re of 1.665/1.655/1.622 Å while the anion shows unequal re of 1.629, 1.787/1.637, 1.958 Å at the B3LYP/PBE0 level and the CCSD(T) level shows equal re of 1.667 Å for all bonds. It has 15 normal modes, viz., 3A1u, 3T2u, 3T2g, 3T1u, 2Eg, 1A1g irreducible representation in the point group symmetry. The 3A1u degenerate modes show zero/imaginary vibrational frequency, while the rest of the 9 normal modes show real frequency but are IR inactive. The IR active T1u mode shows a high IR frequency of 2032/2075/2051 cm−1 with an intensity of 55/63/25 km mol−1. The zero-point vibrational energy is computed to be 9602/10545/8859 cm−1. Thus, it evidences a vibrationally stable HgH6 with Hg6+(5d6) OS while its ions show three/two imaginary frequencies, indicating they are unstable on the PES. An IP of 10.273/10.154/11.694 eV and an EA of 0.550/0.541/−0.273 eV is computed for HgH6. The low/negative EA evidence that the anion is marginally/less stable than the neutral.
3.1.7 HgH7
HgH7 exhibits capped trigonal prismatic molecular geometry with C2V symmetry. It shows three different types of bonds with the re of 1.638, 2.195, 3.613/1.629, 2.417, 3.236 Å along with ∠H1,2HgH3,4 = 90.00/90.04°, ∠H5HgH6 = 90.83/90.40°, and ∠H7HgH8 = 11.79/13.23° at B3LYP/PBE0 level whilst CCSD(T) shows an equal re of 1.715 Å with ∠HHgH = 45°. The long re value at the B3LYP/PBE0 levels suggests that HgH7 is a complex formed between HgH4, H, and H2. The cation shows unequal re of 1.552, 1.650, 1.654, 1.952/1.547, 1.641, 1.643, 1.909 Å at the B3LYP/PBE0 level whilst CCSD(T) shows no convergence. The anion shows an unequal re of 1.656, 1.908, 4.542/1.645, 1.884, 4.307 Å at the B3LYP/PBE0 level whilst CCSD(T) shows an equal re of 1.786 Å for all bonds. The long bond lengths at the B3LYP/PBE0 suggest that anion is also complex formed between HgH40/−, H−/0, and H2. The HgH7 complex shows real frequency while its ions show imaginary frequency due to their unstable nature. An IP of 9.301/9.109/X eV and an EA of 2.375/2.226/2.353 eV is computed for HgH7. The relatively high EA indicates that the complex is stabilized more by an electron capture process similar to HgH5.
3.1.8 HgH8
HgH8 exhibits dodecahedral geometry with D8h symmetry and re of 1.997/1.981/1.795 Å along with ∠HHgH = 45° at the B3LYP/PBE0/CCSD(T) levels suggesting that HgH8 is a molecule with Hg8+(5d4) OS. The removal of an electron results in Hg+…4H2 complex with a large re of 2.701/2.652 Å with negative IP of −2.944/−2.466 eV at the B3LYP/PBE0 level. However, the CCSD(T) level shows a re of 1.743 Å for all bonds indicating it is a stable ion. The negative ion is not converged but it exists as a complex of HgH4−…2H2 moieties at the B3LYP/PBE0 level while the CCSD(T) shows a bond length of 1.843 Å and EA of 3.099 eV. It has 21 normal modes, out of which 10 out of plane angle modes show imaginary frequency and the rest display real frequency. Among the 14 irreducible representations for the D8h symmetry point group viz., A1g, A2g, B1g, B2g, E1g, E2g, E3g, A1u, A2u, B1u, B2u, E1u, E2u, E3u only A2u and E1u are IR active. The IR active E1u mode shows a high IR frequency of 1039/1083 cm−1 with an intensity of 718/739 km mol−1 and zero-point vibrational energy 5802/6082 cm−1 at the B3LYP/PBE0 level.
3.2 Stability of HgHn=1–8
The high valent HgHn molecules are not the global minima but are local minima with a small well depth on the PES. To evaluate their stability, the atomization and the dissociation energy for H2 dissociation channels are calculated and are listed in Table 2.
| Molecule | Dissociation channels | Dissociation energy (eV) |
|---|---|---|
| B3LYP/PBE0/CCSD(T) | ||
| HgH | Hg + H | 0.553/0.543/0.955 |
| HgH2 | Hg + 2H | 3.958/3.890/5.019 |
| Hg + H2 | −0.958/−0.710/0.290 | |
| HgH + H | 3.404/3.347/4.065 | |
| HgH3 |
Hg + 3H Hg + H2 + H HgH + H2 HgH2 + H |
3.984/4.045/4.466 −0.737/−0.555/−0.264 −1.290/−1.098/−1.218 0.222/0.155/−0.554 |
| HgH4 |
Hg + 4H Hg + 2H2 HgH + H2 + H HgH2 + H2 HgH3+ H |
6.919/6.726/9.047 −2.913/−2.474/−0.412 1.450/1.583/3.362 −1.954(−2.073a/−1.763/−0.702) 2.740/2.681/4.581 |
| HgH5 |
Hg + 5H Hg + 2H2 + H HgH +2H2 HgH2 + H2 + H HgH3 + H2 HgH4 + H |
7.035/6.792/9.375 −2.797/−2.408/−0.085 −3.351/−2.951/−1.039 −1.838/−1.700/−0.374 −2.060/−1.853/0.179 0.116/0.066/0.328 |
| HgH6 |
Hg + 6H Hg + 3H2 HgH + 2H2 + H HgH2 + 2H2 HgH3 + H2 + H HgH4 + H2 HgH5 + H |
9.338/9.020/12.595 −5.411/−4.780/−1.594 −1.048/−0.723/2.181 −4.452/−4.070/−1.884 0.242/0.375/3.399 −2.498/−2.306/1.181 2.302/2.228/3.220 |
| HgH7 |
Hg + 7H Hg + 3H2 + H HgH + 3H2 HgH2 + 2H2 + H HgH3 + 2H2 HgH4 + H2 + H HgH5 + H2 HgH6 + H |
11.951/11.410/4.162 −2.797/−2.390/−10.027 −3.351/−2.933/−10.981 −1.839/−1.680/−10.316 2.061/1.835/−9.763 0.116/0.084/−9.614 0.000/0.018/−9.942 2.614/2.390/−8.433 |
| HgH8 |
Hg + 8H Hg + 4H2 HgH + 3H2 + H HgH2 + 3H2 HgH3 + 2H2 + H HgH4 + 2H2 HgH5 + H2 + H HgH6 + H2 |
6.918/6.484/4.034 −12.747/−11.916/−14.885 −8.384/−7.859/−11.110 −11.788/−11.206/−15.175 −7.094/−6.761/−9.891 −9.834/−9.443/−14.472 −5.034/−4.908/−10.070 −5.099/−4.943/−13.291 |
The unimolecular dissociation of hydrogen-rich mercury hydride is unstable for two-body dissociation channels, HgH2 + nH2. However, they are stable with positive dissociation energy for H atom reactions, and the atomization energy increases systematically and reaches a maximum for HgH7 and drops for HgH8. It indicates that they are meta-stable and can survive if the transition state (TS) barrier is sufficiently high on the PES for its preparation and identification by low-temperature matrix isolation or supersonic molecular beam techniques. Therefore, the TS of HgH3, HgH4, and HgH6 are computed and are given in Figures 1-3. The ZPVE corrected values are 0.056/0.058, 0.421/0385, and 0.337/0.201 eV for HgH3, HgH4, and HgH6 at the B3LYP/PBE0 levels. A TS with barrier heights of 0.404 and 0.311 eV for HgH4 and HgH6 has been reported earlier at the B3LYP level and agrees well with the values obtained here. Moreover, HgH3 shows a high-energy isomer at 0.859/0.716 eV above the ground state. It should be noted that the HArF compound with a barrier height of 0.477 eV was synthesized and detected in a solid argon matrix at 7.5K [56-59], indicating that it may also be plausible to prepare and characterize HgH4 and HgH6 using the matrix isolation technique.



In addition to the dissociation energy and TS, the HOMO–LUMO energy gap is also computed as high HOMO–LUMO gap indicates high kinetic stability and low chemical reactivity because the addition of an electron to LUMO, and the removal of an electron from HOMO is energetically unfavorable [60-63]. Figures 4-6 show the electron density, bonding molecular orbitals, HOMO–LUMO plot, and their energy gaps for HgH2, HgH4, HgH6, and HgH8. A large HOMO–LUMO energy gap of 6.827/7.279, 6.914/7.437, 6.944/7.039 eV for HgH2, HgH4, HgH6, and a relatively low 3.404/3.439 eV for HgH8 at the B3LYP/PBE0 level. The HOMO–LUMO gap of HgH4 and HgH6 is higher than the energy gap of experimentally detected HgF4 (4.556/5.317 eV) and HgF6 (4.185/5.015 eV) [19] and supports the existence of stable high valent HgHn.



3.3 Chemical Bonding and Population Analysis
Molecules are best described by the collection of nuclei surrounded by electrons, and the electron distributions reflect the nature and strength of bonds between the atoms in molecules. The change in the electron density distribution from the simple sum of atom density provides insight into the nature and strength of bonds in molecules. Quantum chemical methods provide atomic charge redistribution in the molecular system based on the population analysis or partitioned scheme. The natural population analysis and local properties computed by the LoProp approach are less basis set dependent than the Mulliken population analysis schemes. Therefore, both schemes are employed to calculate the charge distribution characteristics of HgHn. Table 3 lists the charge accumulation on Hg and H by Mulliken and LoProp schemes. Figure 7a shows variation in Mulliken charge on Hg for each H atom with a maximum at HgH8 and relatively less at HgH6. The charge accumulation trends are not in alignment with the HOMO–LUMO gap. In contrast, LoProp charge analysis shows a gradual increase in charge on Hg for each added H atom with a maximum at HgH6 and then a decrease in HgH8, and truly reflects the HOMO–LUMO gap of HgHn. Furthermore, LoProp charge accumulation on Hg in HgHn and HgFn computed with the ANO-RCC-VQZP basis set at the PBE0 level is shown in Figure 7b. It indicates that the magnitude of the charge on Hg is high in HgFn and relatively low in HgHn, but both systems display the same trend and truly reflect the HOMO–LUMO gap in both systems. Thus, both the charge distribution and natural bond order analysis support the existence of high OS in HgIVH4 (d8), HgVIH6(d6), and HgVIIIH8(d4). However, the odd electron species shows a nonuniform charge distribution, indicating complex formation by noncovalent interaction between the precursor HgHn = 2,4,6 and H/H2, respectively.
| Charge | HgH2 | HgH3 | HgH4 | HgH5 | HgH6 | HgH7 | HgH8 | |
|---|---|---|---|---|---|---|---|---|
| Mulliken | QHg | 0.4274/0.5499 | 0.4433/0.5961 | 0.5640/0.7873 | 0.5259/0.7351 | 0.5578/0.8113 | 0.5109/0.7042 | 0.8694/1.1366 |
| qH1 | −0.2137/−0.2750 | −0.1975/−0.2562 | −0.1410/−0.1968 | −0.1318/−0.1867 | −0.0929/−0.1352 | −0.1309/−0.1826 | −0.1087/−0.1422 | |
| qH2 | −0.2137/−0.2750 | −0.1975/−0.2562 | −0.1410/−0.1968 | −0.1318/−0.1867 | −0.0929/−0.1352 | −0.1309/−0.1826 | −0.1087/−0.1422 | |
| qH3 | −0.0483/−0.0838 | −0.1410/−0.1968 | −0.1318/−0.1867 | −0.0929/−0.1352 | −0.1309/−0.1826 | −0.1087/−0.1422 | ||
| qH4 | −0.1410/−0.1968 | −0.1318/−0.1867 | −0.0929/−0.1352 | −0.1309/−0.1826 | −0.1087/−0.1422 | |||
| qH5 | 0.0015/0.0117 | −0.0929/−0.1352 | 0.0027/0.0043 | −0.1087/−0.1422 | ||||
| qH6 | −0.0929/−0.1352 | 0.0027/0.0043 | −0.1087/−0.1422 | |||||
| qH7 | 0.0075/0.0175 | −0.1087/−0.1422 | ||||||
| qH8 | −0.1087/−0.1422 | |||||||
| LoProp | QHg | 0.7541/0.7607 | 0.8906/0.8922 | 1.2418/1.2421 | 1.2723/1.2479 | 1.5843/1.5744 | 1.2699/1.2483 | 1.1797/1.1866 |
| qH1 | −0.3770/−0.3804 | −0.3699/−0.3719 | −0.3104/−0.3105 | −0.3074/−0.3092 | −0.2640/−0.2624 | −0.3076/−0.3089 | −0.1475/−0.1483 | |
| qH2 | −0.3770/−0.3804 | −0.3699/−0.3719 | −0.3104/−0.3105 | −0.3074/−0.3092 | −0.2640/−0.2624 | −0.3076/−0.3089 | −0.1475/−0.1483 | |
| qH3 | −0.1509/−0.1484 | −0.3104/−0.3105 | −0.3074/−0.3092 | −0.2640/−0.2624 | −0.3076/−0.3089 | −0.1475/−0.1483 | ||
| qH4 | −0.3104/−0.3105 | −0.3074/−0.3092 | −0.2640/−0.2624 | −0.3076/−0.3089 | −0.1475/−0.1483 | |||
| qH5 | −0.0426/−0.0110 | −0.2640/−0.2624 | −0.0016/−0.0090 | −0.1475/−0.1483 | ||||
| qH6 | −0.2640/−0.2624 | −0.0016/−0.0090 | −0.1475/−0.1483 | |||||
| qH7 | −0.0363/−0.0019 | −0.1475/−0.1483 | ||||||
| qH8 | −0.1475/−0.1483 | |||||||
| Natural Bond Order | 3.983 electrons in 2 bonds |
NO HgH bond 0.920 non-bonded electrons on Hg, 1.014 non-bonded electrons on H, and the remaining 5.144 electrons are diffuse. |
7.901 electrons are involved in 4 bonds. |
7.872 electrons in 4 bonds and 0.963 non-bonded electrons on H5 |
11.859 electrons in 6 bonds |
9.976 electrons are involved in 5 bonds, and the remaining 1.214 electrons are diffuse. | 11.744 electrons in 6 bonds, others did not converge. |

The above structure, stability, charge redistribution, and chemical bonding data analysis indicate that HgHn ≥ 3 is stable and can be prepared only at very low temperatures by the reaction of the Hg atom in an excited state with H2/D2. The reaction products can be identified and characterized using matrix isolation IR spectroscopy supported by high-level quantum chemical calculations for their spectroscopic signature, structural, and spectroscopic data. However, it is significant to note that the HgH2 prepared by the matrix isolation technique doesn't truly reflect the formal oxidation state of Hg, as Hg is generated from the excited state (3P1) rather than in the ground state (1S0) during the reaction.
Recently, HgH2/HgD2 were synthesized through a direct gas phase reaction between Hg vapor and H/D atoms obtained from H2/D2 electrical discharge. The analysis of vibration-rotation transitions in the reaction products confirms the formation of HgH2/HgD2 with Hg in OS II [47]. Similarly, high valent HgHn ≥ 3 can also be synthesized by the same technique. The vibrational-rotational spectral analysis by a high-resolution FTIR spectrometer may provide direct evidence for the existence of high valent molecules, as the HOMO–LUMO gap values of HgH4 and HgH6 are similar to HgH2 and confirm the direct participation of d shell electrons in chemical bonding and the influence of the special theory of relativity in Hg. Alternatively, direct expansion of Hg vapor and the H atoms obtained from the electric discharge of H2 in a supersonic beam source can be used for their preparation, followed by electronic spectroscopy like laser absorption/laser-induced fluorescence/photoionization/mass-selected photoelectron spectroscopy characterization. However, there is ambiguity in identifying the high-valent species by using IR frequencies computed by various theoretical methods. Therefore, alternative methods other than conventional vibrational spectroscopy signatures are sought after. Historically, the behavior of atoms and molecules in electric (Stark effect) and magnetic fields (Zeeman splitting) has been investigated to understand the quantum structure of atoms and molecules in depth. Hence, the behavior of HgHn under the influence of an electric field has been studied to help their unambiguous detection using Stark spectroscopy/molecular beam deflection or matter-wave interferometry techniques.
The LoProp method in MOLCAS partitions the molecular polarizability into localized polarizabilities that consist of dipole and charge mobility contributions using finite field perturbation theory (FFPT) [64, 65]. Accordingly, the change in charge due to the external electric field (EEF) is given by Δq = ΔP(AB). ξ(AB) where ΔP(AB) represents the difference in the electrostatic potential and ξ(AB) measures the ability to conduct charge in a bond for a fixed potential difference between the bonded atoms (bond/charge capacitance). The bond capacitance magnitude helps to classify different types of bonds that correlate well with bond order, polarity, conjugation, and electronegativity, and holds chemical information. Further, the bond capacitance reflects the chemical properties remarkably well in various organic molecules with different bond orders and metal-containing molecules [66, 67]. Figure 8 illustrates that the HgH bond capacitance in HgHn gradually increases, showing a maximum for HgH6 and declines for HgH8 at a low field strength of 0.001 a.u. and echo the HOMO–UMO gap and LoProp charge on Hg atom. The ξ(AB) value for the HgH bond is 1.8, 2.0, 2.1, and 1.5 for HgH2, HgH4, HgH6, and HgH8, respectively, which is comparable to the C=C bond value (ξ(AB) = 1.923) found in benzene.

To assist their detection and identification using Stark spectroscopy/molecular beam deflection technique. We have computed the behavior of HgH bond conductance under varying applied fields. The field strength ranges from 0.001 to 1 a.u. (1a.u. = 514.225 V nm−1) is shown in Figure 9. The ξ(AB) value shows a linear dependence up to a field strength of 0.1 a.u. The values gradually decrease for increasing field strength indicating that 0.1 a.u. is the optimum field strength for their detection using molecular beam deflection or the matter wave interferometry technique [68-75]. Further details regarding the computations, as well as the HgH bond conductance and isotropic polarizability of HgH3 and HgH5 are given in the Supporting Information S1.

4 Conclusion
The structure, stability, bonding, electronic properties, and electrical characteristics of the HgHn=1–80,± were investigated by DFT-B3LYP/PBE0 and CCSD(T) methods. The HgH4, HgH6, and HgH8 molecules exhibit high HOM-LUMO gaps of 6.914/7.437 eV, 6.944/7.039 eV, and 3.404/3.439 eV with a barrier height of 0.421/0385 eV and 0.337/0201 eV for HgH4 and HgH6. The HgH bond capacitance (ξ(AB)) in the HgHn increases gradually with size and then exhibits a maximum at HgH6 and a decline for HgH8. The HgH bond capacitance ranges from 1.52 to 2.09 and is comparable to the C=C bond capacitance value (ξ(AB) = 1.923) in benzene, evidencing a stable high-valent HgHn and exhibiting high oxidation states of Hg ranging from I to VIII in HgHn. Furthermore, the polarizability and ξ(AB) demonstrate a linear response to external electric fields up to 0.1 a.u. for their potential detection using molecular beam deflection or matter-waver interferometry techniques.
Acknowledgments
The author would like to thank Mr. Vaibhav Kumar, kum. Duggal Vibhuti, and Mr. Kislay Bhatt from the Supercomputing Facility of Computer Division, BARC for their generous computer time allocation and support.
Conflicts of Interest
The author declares no conflicts of interest.
Open Research
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.

