

Journal list menu

 Issue
IssueClinical Genetics: Volume 105, Issue 2
113-230February 2024
Export Citations
Download PDFs
ISSUE INFORMATION
REVIEWS
Association between genetic variants of transmembrane transporters and susceptibility to anthracycline-induced cardiotoxicity: Current understanding and existing evidence
- Pages: 115-129
- First Published: 14 November 2023

The majority of single nucleotide polymorphisms (SNPs) associated with anthracycline pharmacogenomics were identified in the ATP-binding cassette (ABC) and solute carrier (SLC) transporters, generating increasing interest in the pharmacogenetic implications of their genetic variations for anthracycline-induced cardiotoxicity (AIC). The asterisk (*) highlights the extensively studied genetic variant associated with anthracycline transport. Specifically, the SLC28A3 rs7853758 variant has been employed in clinical practice to predict the risk of developing AIC.
Gene mutations as predictors of central lymph mode metastasis in cN0 PTC: A meta-analysis
- Pages: 130-139
- First Published: 20 November 2023
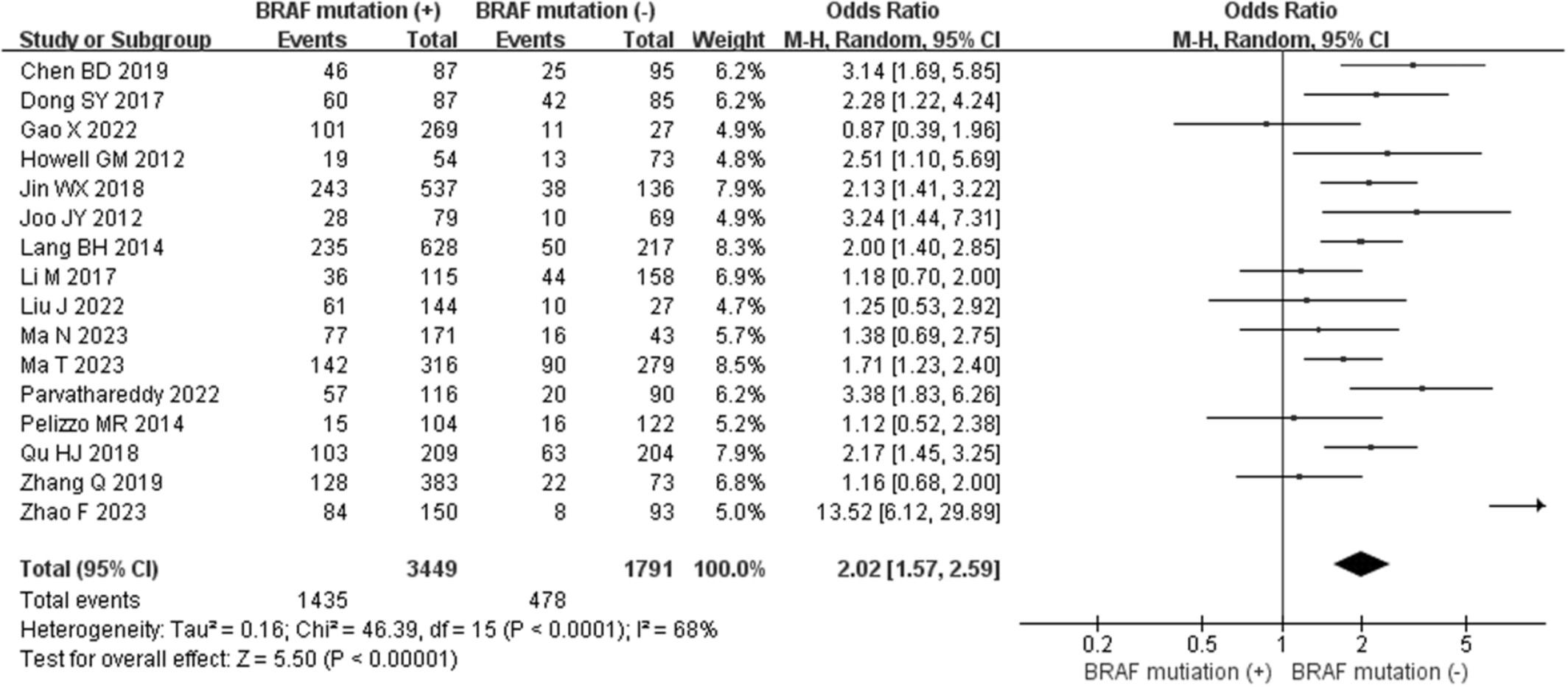
We found BRAF mutation was a predictive factor for CLNM in cN0 PTC, as well as cN0 PTMC, which could be useful for clinician to accurately choose phylactic CLND and better manage patients with cN0 PTC or PTMC. More prospective studies are needed to validate the findings of our study.
ORIGINAL ARTICLES
Genetic and phenotypic findings in 34 novel Spanish patients with DDX3X neurodevelopmental disorder
- Pages: 140-149
- First Published: 30 October 2023
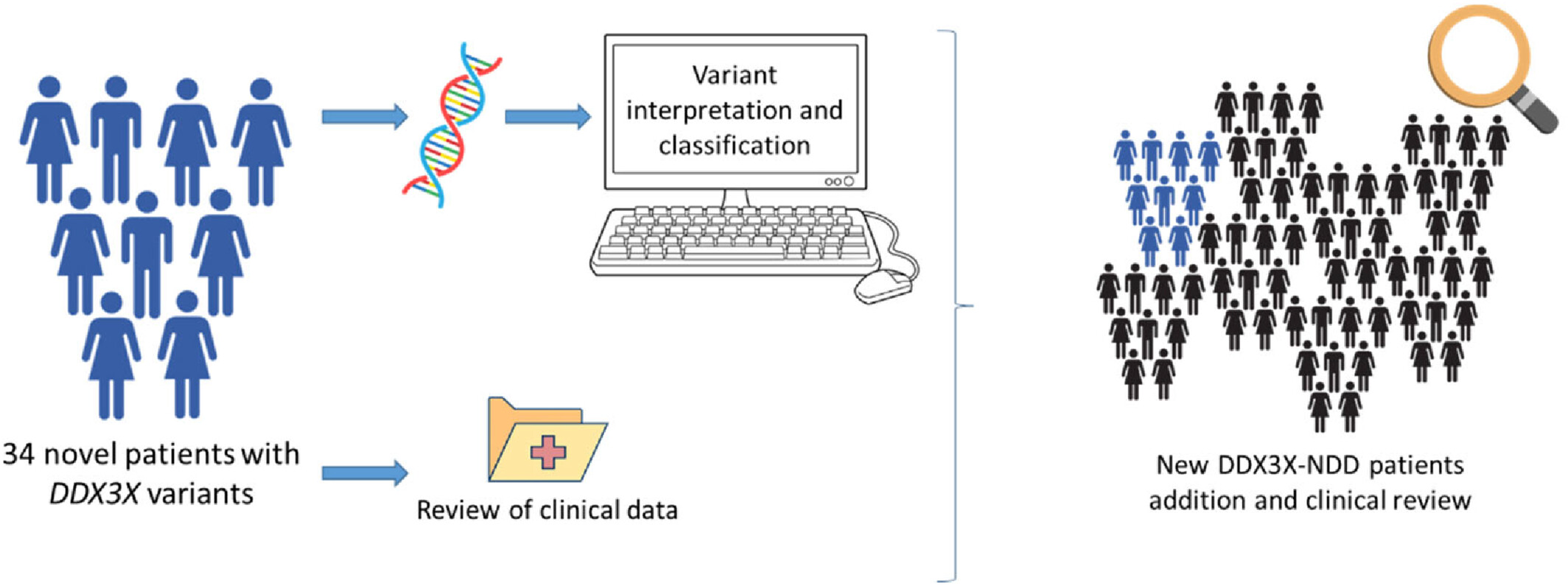
In this report we describe 34 new patients with a variant in DDX3X and reviewed 200 additional patients previously reported in the literature, contributing with 25 novel variants and a deep phenotypic characterization.
Perspectives of carriers of X-linked retinal diseases on genetic testing and gene therapy: A global survey
- Pages: 150-158
- First Published: 20 October 2023
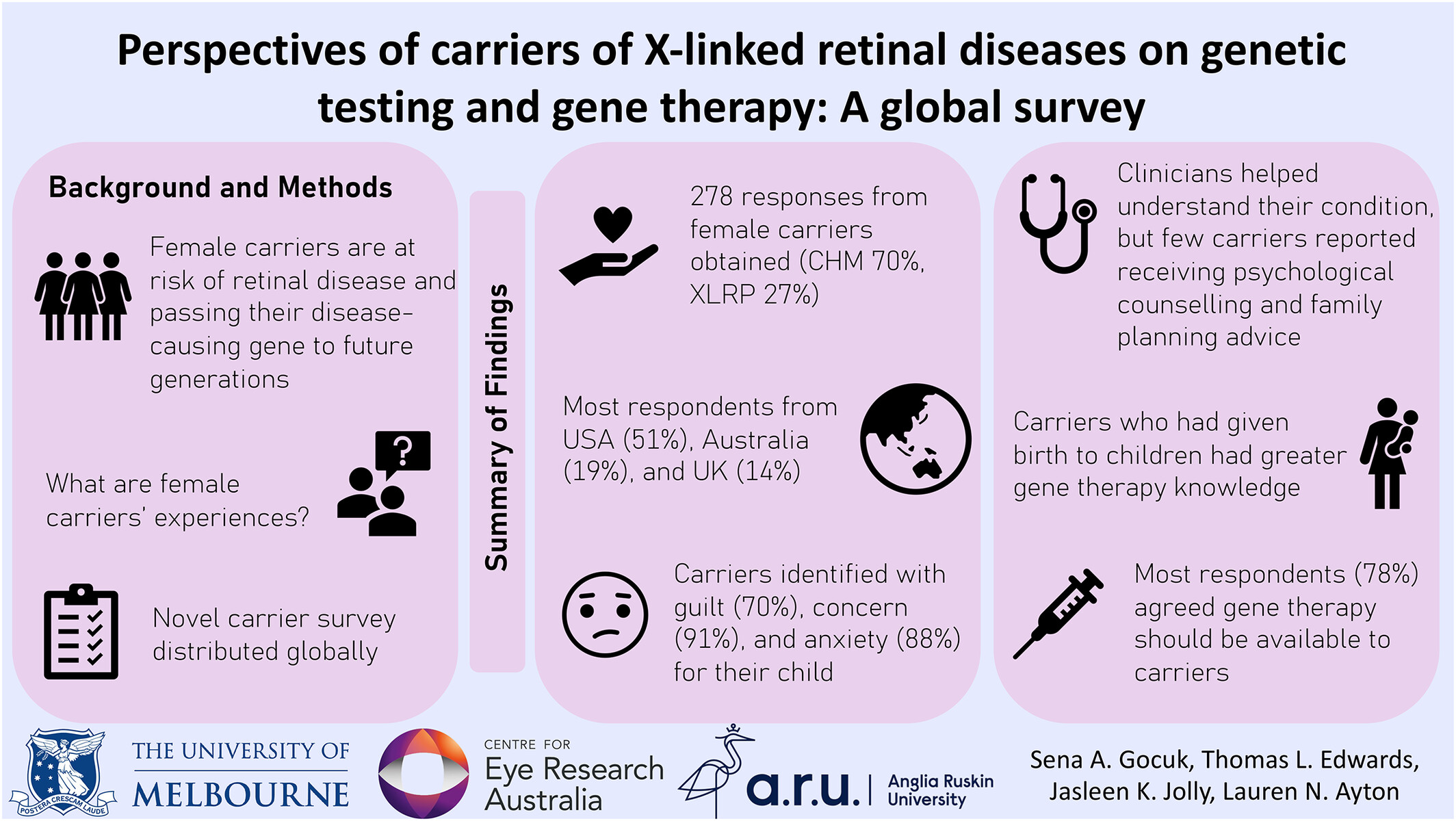
Self-reported female carriers of X-linked retinal disease genetic testing experiences, emotions, and knowledge relating to genetic testing, genetic counselling, and gene therapy.
Rigorous evaluation of genetic and epigenetic effects on clinical laboratory measurements using Japanese monozygotic twins
- Pages: 159-172
- First Published: 29 October 2023
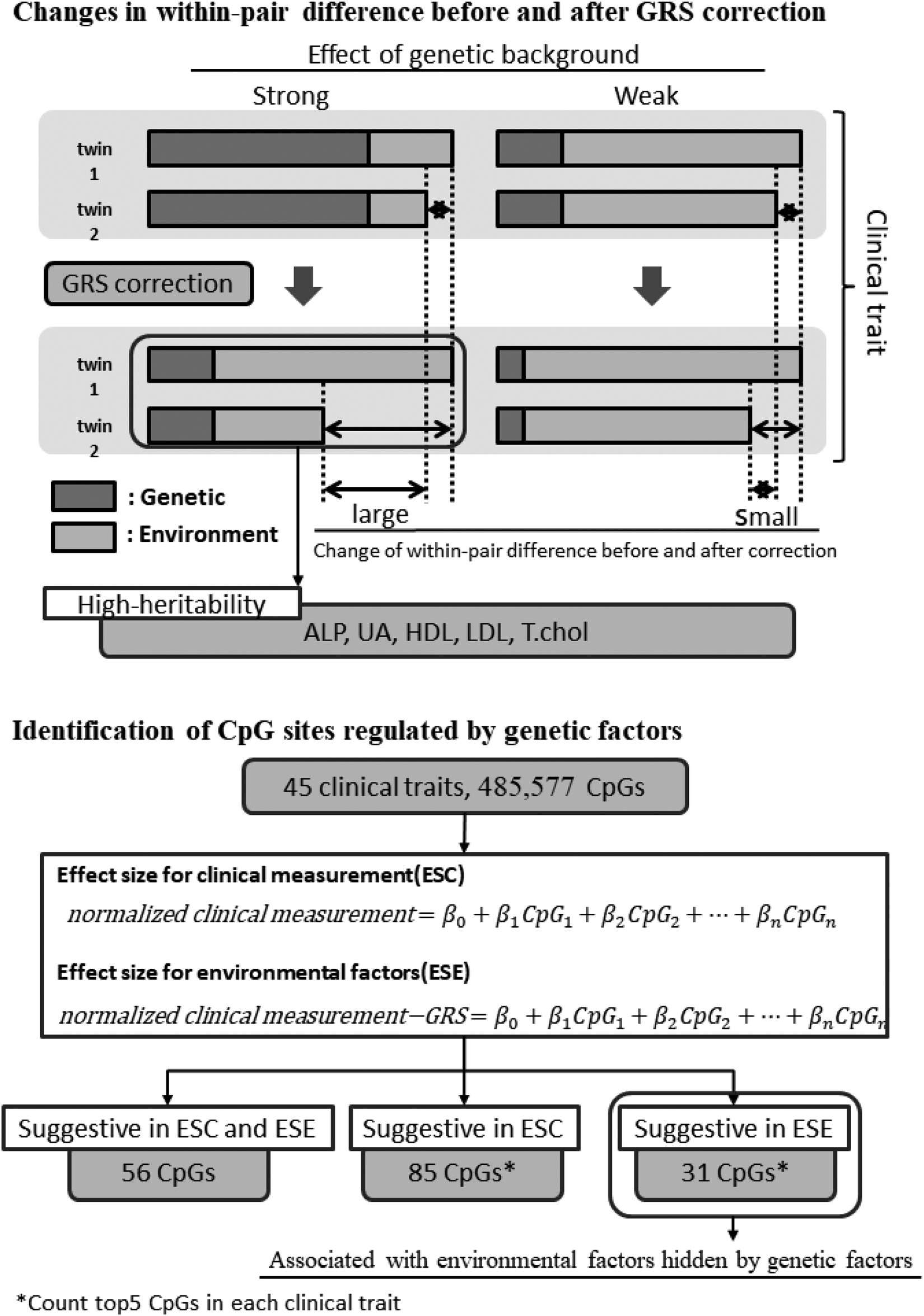
Monozygotic twin samples showed that five clinical measurements were strongly genetically affected. CpG sites are classified into three groups: regulated only by environmental factors, by environmental factors masked by genetic factors, and only by genetic factors. Our method has the potential to identify trait-related methylation sites.
Clinical findings in individuals with duplication of genes associated with X-linked intellectual disability
- Pages: 173-184
- First Published: 29 October 2023
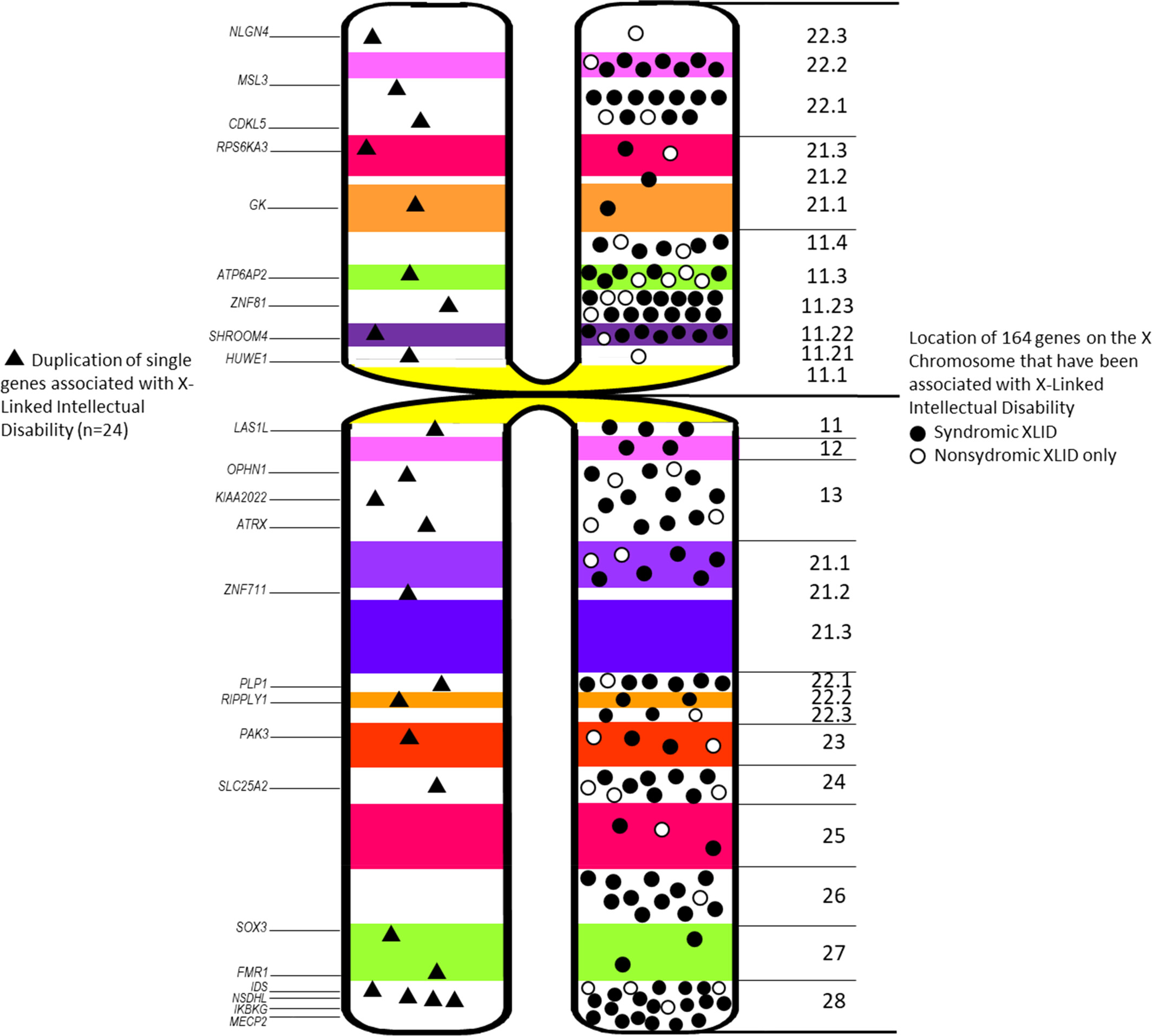
Location of single XLID gene duplications (left) and location of all 164 XLID genes (right).
SHORT REPORTS
DNA-pools targeted-sequencing as a robust cost-effective method to detect rare variants: Application to dilated cardiomyopathy genetic diagnosis
- Pages: 185-189
- First Published: 30 October 2023
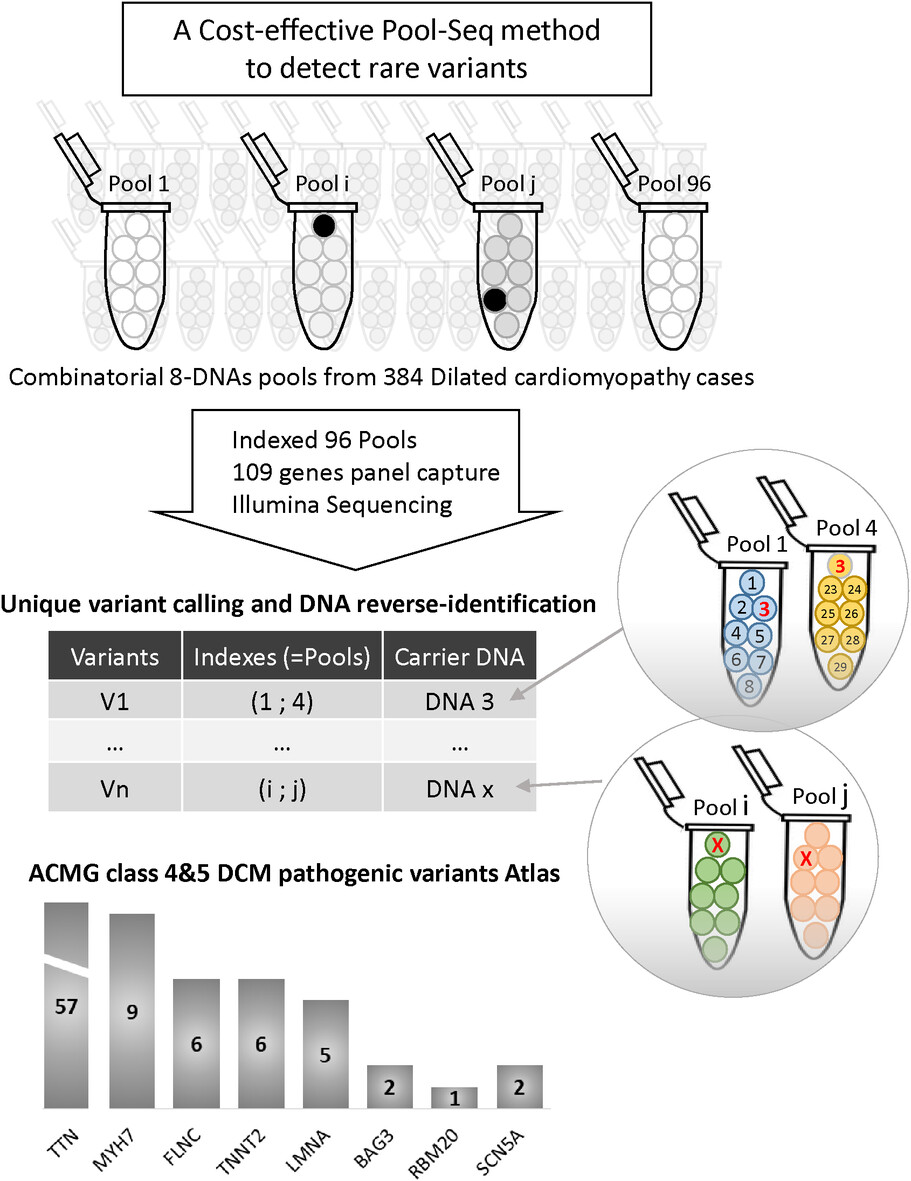
We report an original pool-sequencing NGS method accurately detecting rare variants in 384 dilated cardiomyopathy cases. This innovative approach is cost-effective for genetic diagnostic in rare diseases.
A novel non-recurrent CNV deletion involving TBX4 and leaving TBX2 intact causes congenital alveolar dysplasia
- Pages: 190-195
- First Published: 11 October 2023
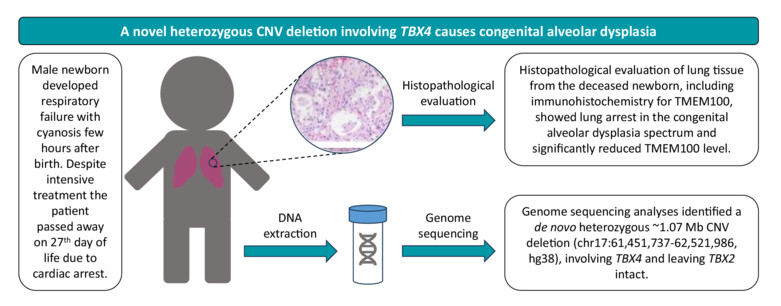
We describe a male neonate who died at 27 days of life from acute respiratory distress caused by lung growth arrest along the spectrum of congenital alveolar dysplasia confirmed by histopathological assessment. Trio-based genome sequencing revealed in the proband a de novo heterozygous ~1.07 Mb CNV deletion at 17q23.2, encompassing TBX4.
Adding to the evidence of gene-disease association of RAP1B and syndromic thrombocytopenia
- Pages: 196-201
- First Published: 18 October 2023
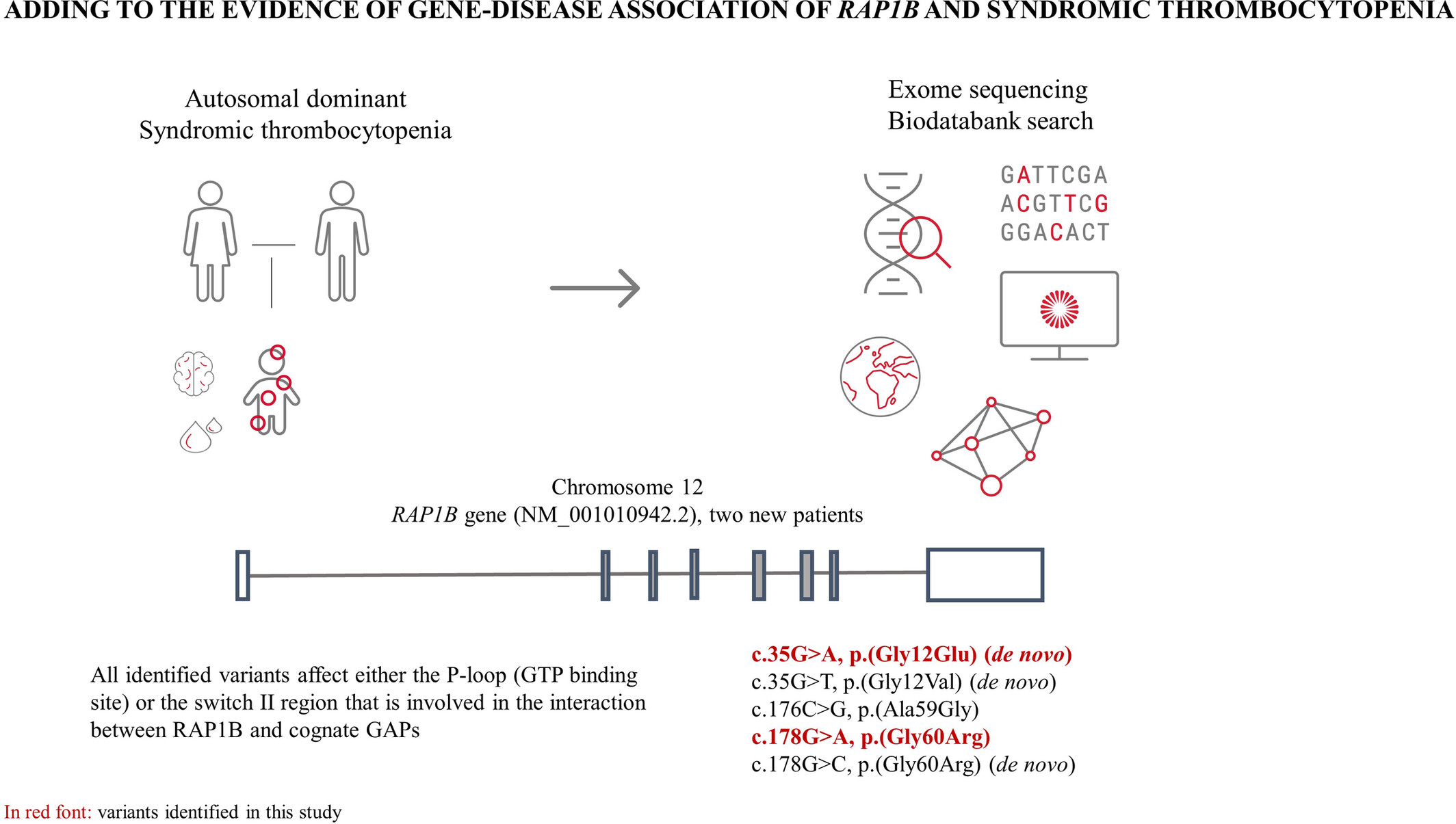
All identified variants affect either the P-loop (GTP binding site) or the switch II region that is involved in the interaction between RAP1B and cognate GAPs.
A homozygous frameshift variant expands the clinical spectrum of SAMD9 gene defects
- Pages: 202-208
- First Published: 13 October 2023
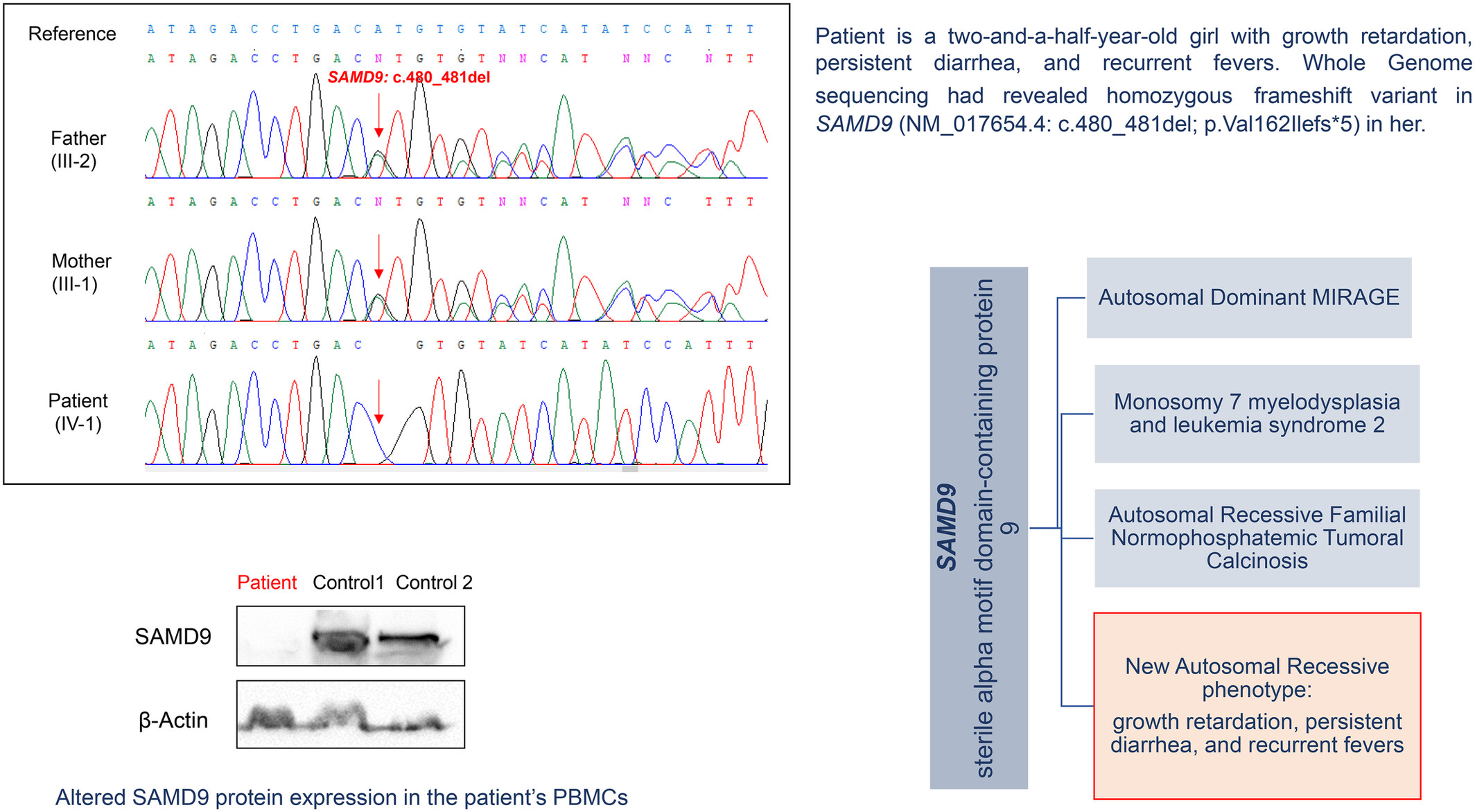
Expanding the clinical spectrum of SAMD9-linked diseases. A homozygous frameshift variant in SAMD9 (NM_017654.4: c.480_481del; p.Val162Ilefs*5) was identified in a two-and-a-half-year-old girl with growth retardation, persistent diarrhea, and recurrent fevers.
The third case of Marbach-Rustad progeroid syndrome caused by a de novo LEMD2 variant
- Pages: 209-213
- First Published: 23 October 2023
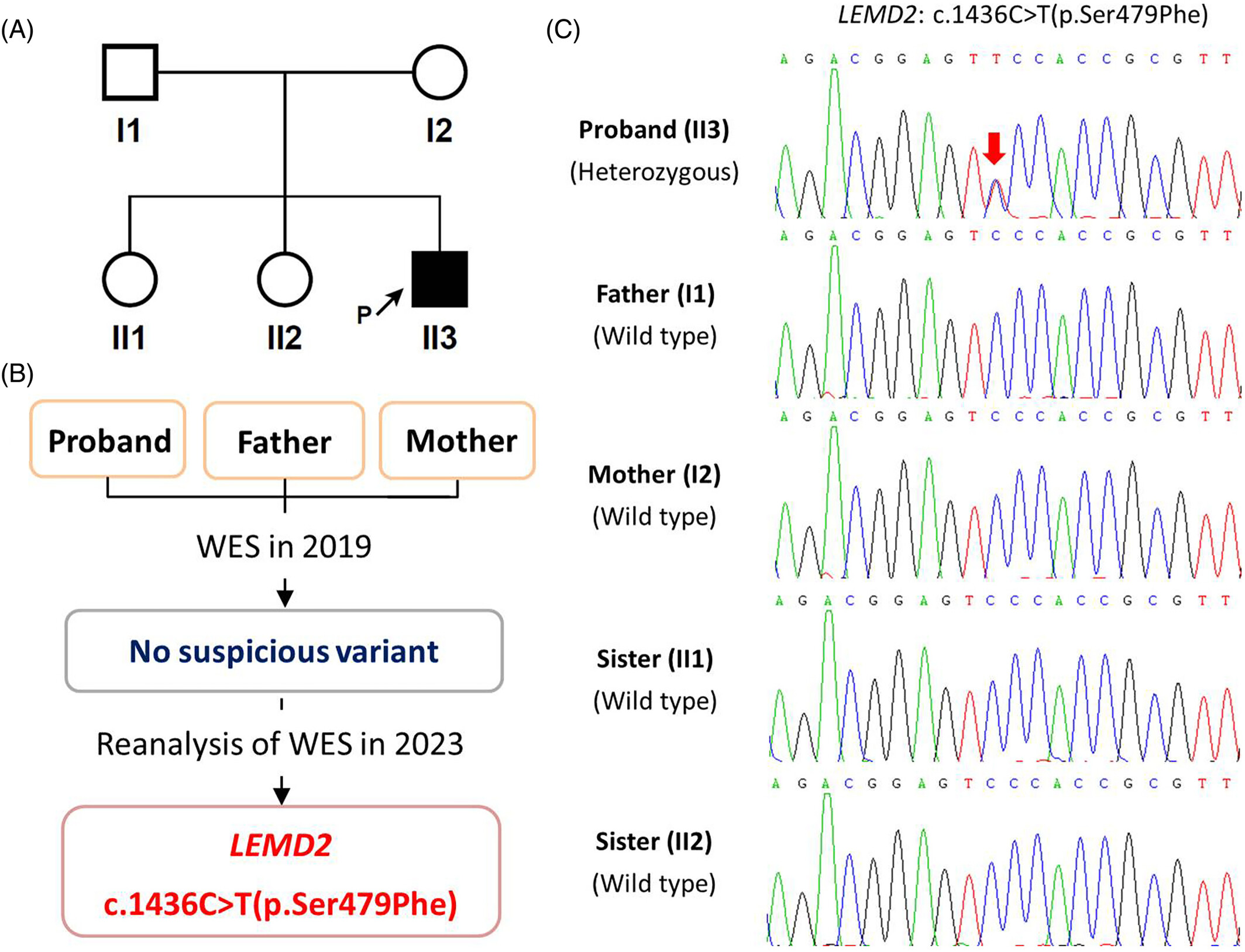
The third case of Marbach-Rustad progeroid syndrome. (A) The family pedigree. (B) Whole exome sequencing (WES) in 2019 did not find a suspicious variant, whereas reanalysis in 2023 revealed LEMD2 c.1436C>T(p.Ser479Phe) variant as the cause. (C) Sanger sequencing confirmed the de novo LEMD2 c.1436C>T(p.Ser479Phe) heterozygous variant in the proband.
De novo enhancer deletion of LMX1B produces a mild nail-patella clinical phenotype
- Pages: 214-219
- First Published: 29 October 2023
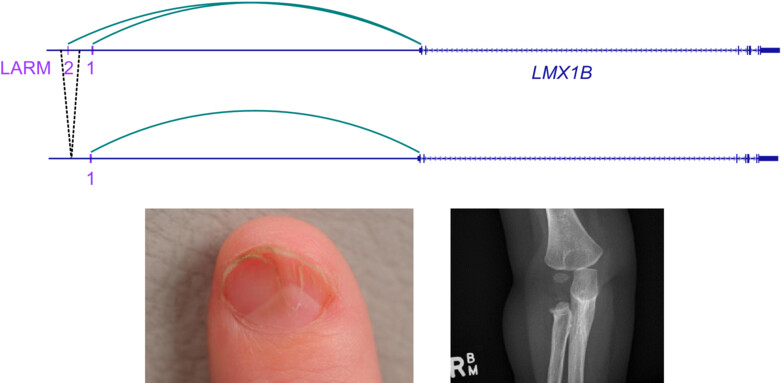
The Nail-Patella syndrome gene, LMX1B, is regulated by two upstream elements, LARM1 and 2. Deletion of LARM2 in multiple-affected family members is associated with a milder form of this syndrome, characterised by dysplastic nails and smaller patellae.
A splice donor variant of GAS8 induces structural disorganization of the axoneme in sperm flagella and leads to nonsyndromic male infertility
- Pages: 220-225
- First Published: 11 November 2023
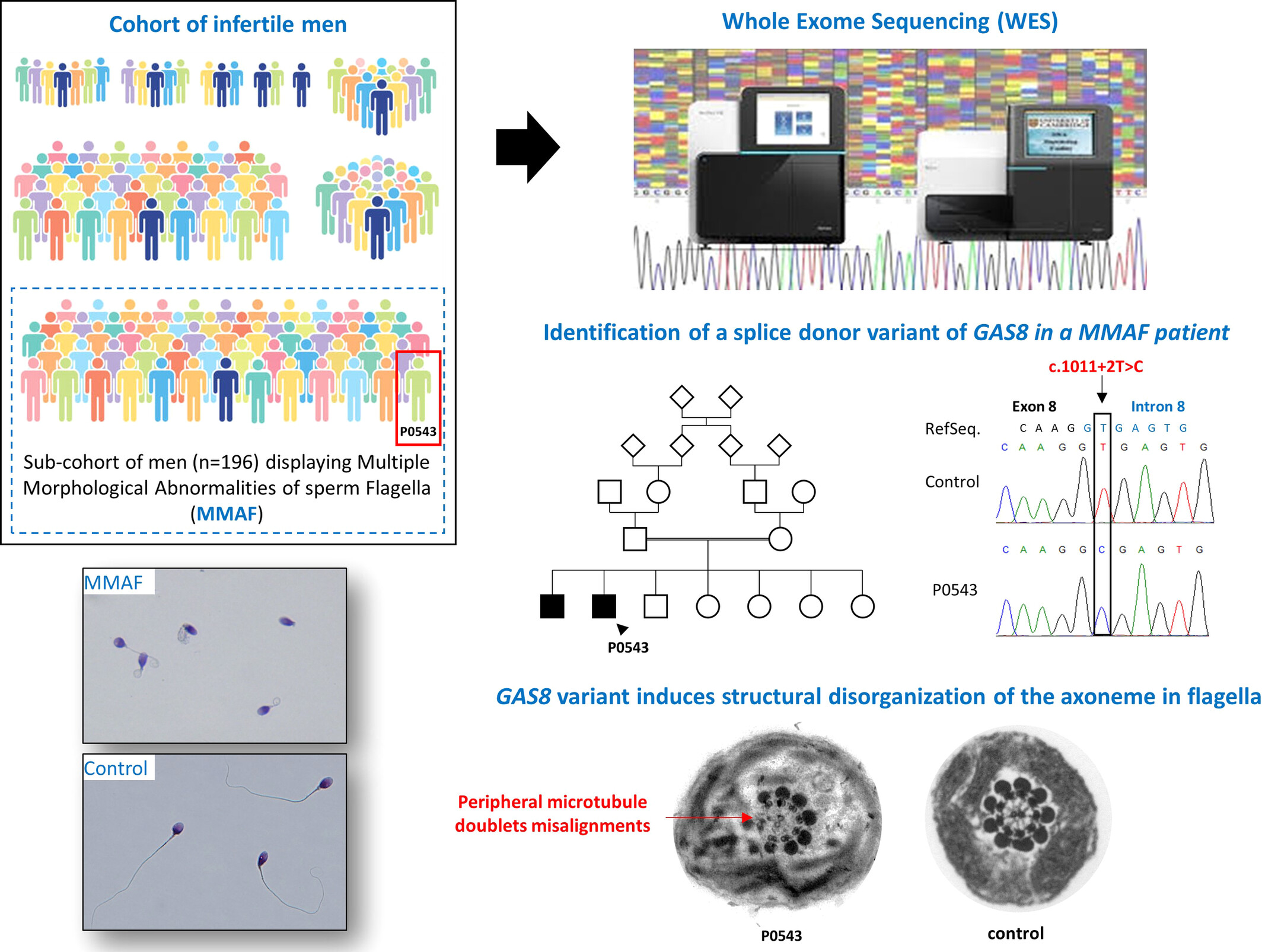
From a large cohort of infertile men, we focused on a subcohort of 196 men displaying asthenozoospermia due to multiple morphological abnormalities of the sperm flagella (MMAF). Whole exome sequencing (WES) data analysis allowed the identification of rare homozygous splicing variant in GAS8, disrupting the nexin-dynein regulatory complex (N-DRC) and inducing a severe disorganisation of the axoneme in sperm flagella.
RESEARCH LETTERS
Variants in DOK7 results in fetal akinesia deformation sequence: A case report and review of literature
- Pages: 226-227
- First Published: 17 October 2023
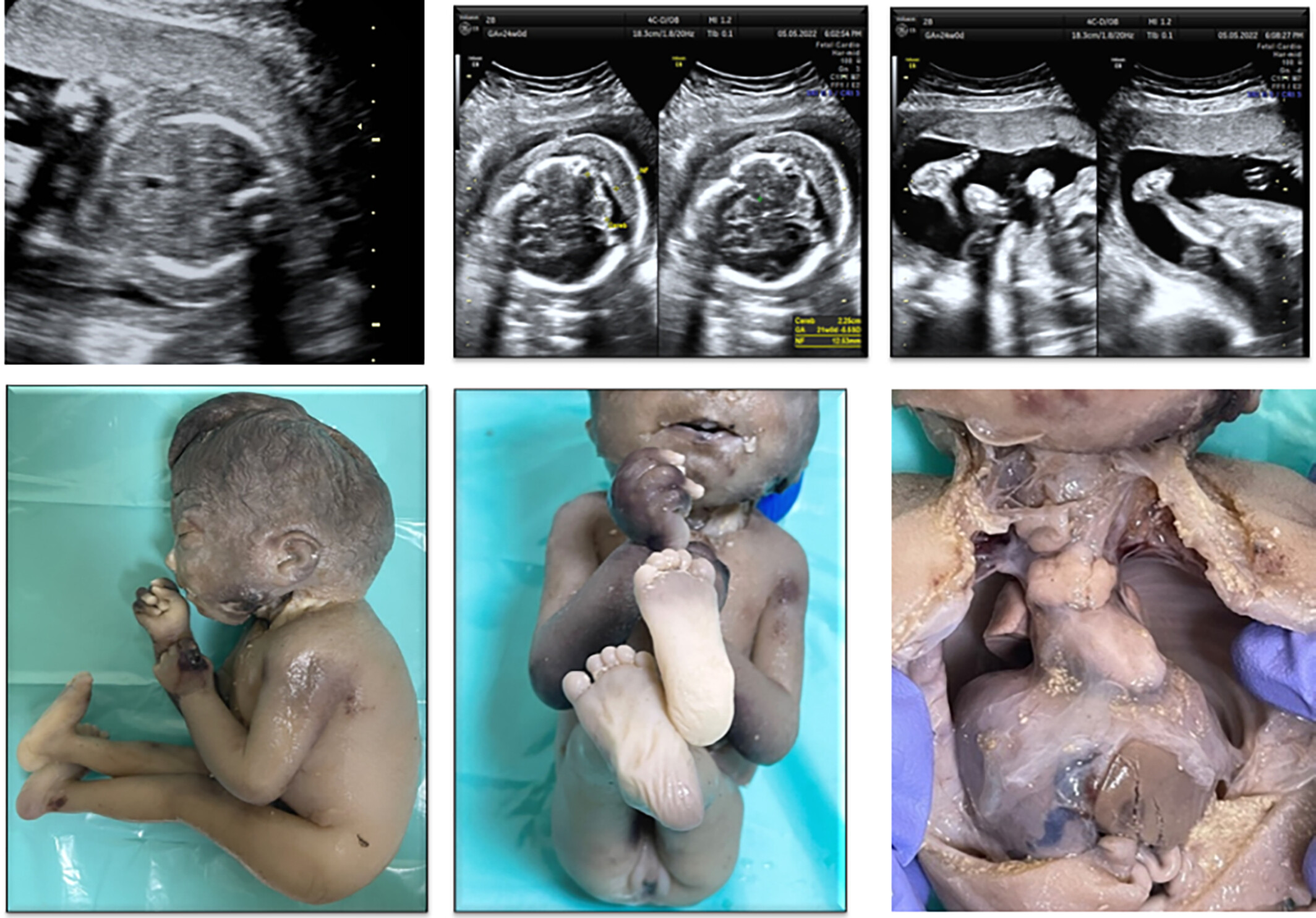
We report the third case of FADS due to biallelic DOK7 variants, which further strengthens the association of DOK7 with this lethal phenotype and lack of genotype phenotype correlation.
Progressive ataxia, ophthalmoparesis, and hypogonadotropic hypogonadism in a family with a novel variant in the KIFBP gene
- Pages: 228-230
- First Published: 30 October 2023
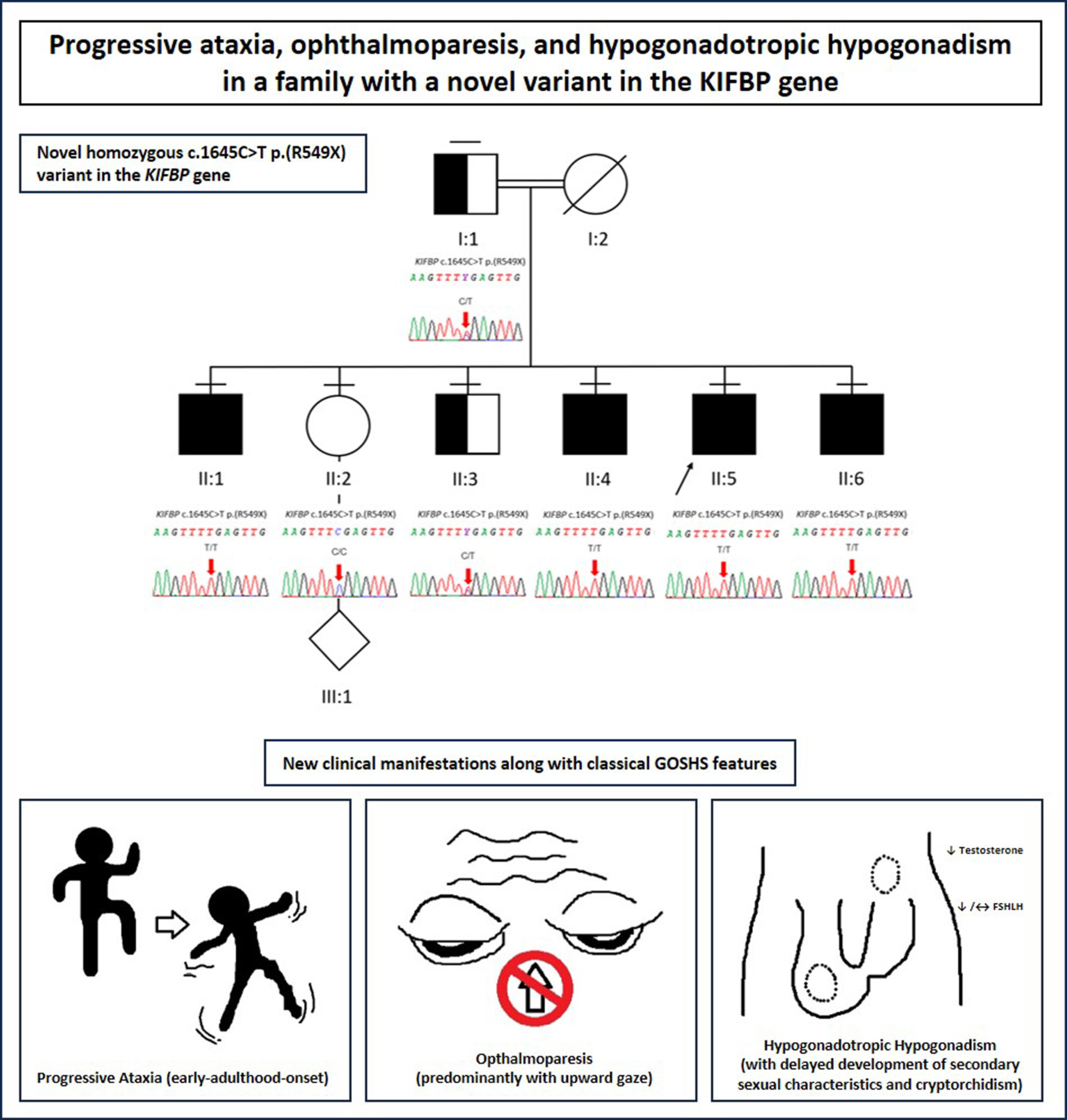
A novel homozygous variant in KIFBP was identified in a consanguineous family with four sibs affected by Goldberg–Sphrintzen Syndrome (GOSHS). We report for the first time, early-adulthood-onset progressive ataxia, opthalmoparesis, and hypogonadotropic hypogonadism in GOSHS.