

Journal list menu


Export Citations
Download PDFs
Cover Image, Volume 38, Issue 9
- Page: i
- First Published: 17 August 2017

On the cover: This cover image, by Qifang Xu et al., is based on the Special Article Benchmarking predictions of allostery in liver pyruvate kinase in CAGI4, Pages 1123–1131. DOI: 10.1002/humu.23222.
Issue Information
- Pages: 1033-1035
- First Published: 17 August 2017
Novel ID gene CSNK2B: The crossover from molecular diagnosis to research continues
- Page: 1037
- First Published: 17 August 2017
Reports from CAGI: The Critical Assessment of Genome Interpretation
- Pages: 1039-1041
- First Published: 12 July 2017
Performance of in silico tools for the evaluation of p16INK4a (CDKN2A) variants in CAGI
- Pages: 1042-1050
- First Published: 25 April 2017
The Critical Assessment of Genome Interpretation (CAGI) experiment is aimed to define the state of art of genotype-phenotype interpretation. Here, we present the assessment of the CAGI p16INK4a challenge. Participants were asked to predict the effect on cellular proliferation of ten variants for the p16INK4a tumor suppressor, a kinase inhibitor coded by the CDKN2A gene. Twenty-two pathogenicity predictors were validated in terms of accuracy and reliability. Different assessment measures were combined in an overall ranking to provide robust results.
Assessing predictions of fitness effects of missense mutations in SUMO-conjugating enzyme UBE2I
- Pages: 1051-1063
- First Published: 17 August 2017
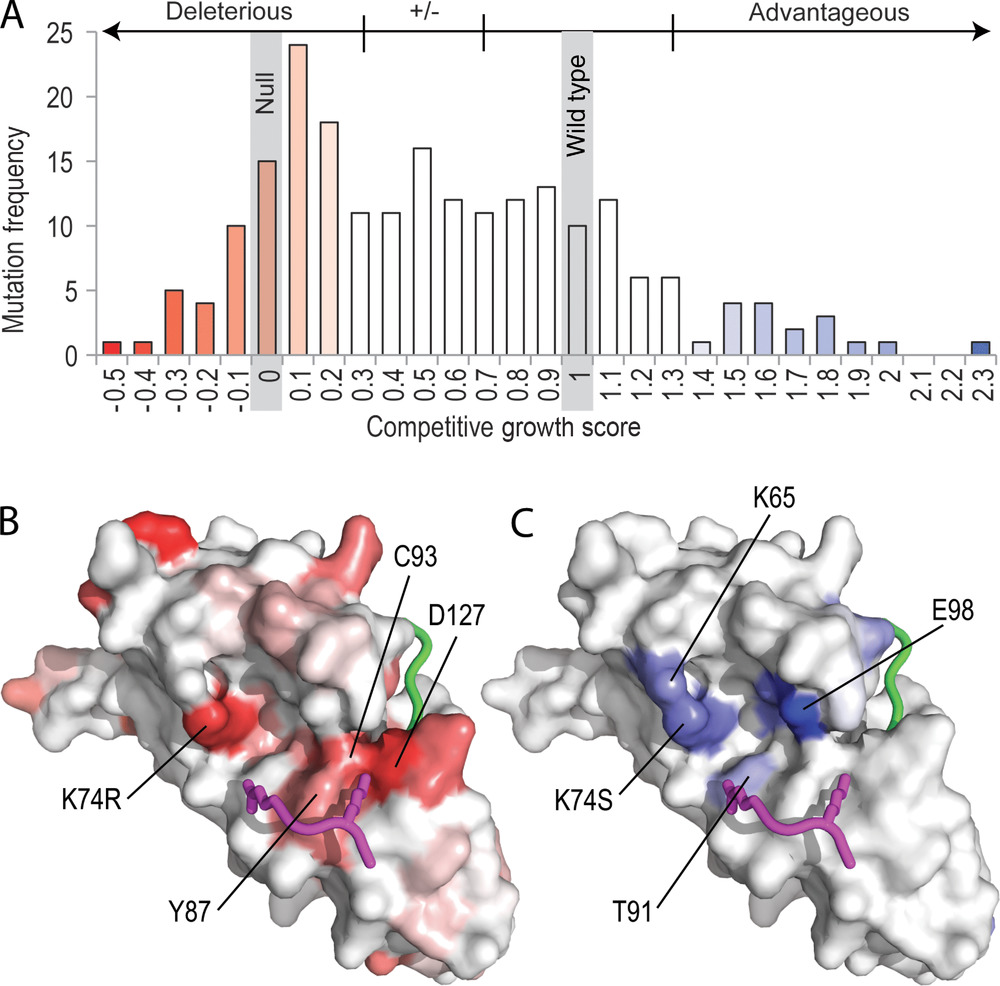
Competitive Growth Scores of UBE2I Mutations. A) A histogram of scores. Bars are colored in gradient from red (deleterious mutations) through white (wild type) to blue (advantageous mutations). The same color gradient is applied to corresponding residues in surface representations of the UBE2I structure for B) deleterious and C) advantageous mutations. The consensus substrate tetrapeptide PsiKxE (magenta) and the C-terminal SUMO peptide (green) are shown and select residues near the active site are labeled.
Blind prediction of deleterious amino acid variations with SNPs&GO
- Pages: 1064-1071
- First Published: 19 January 2017
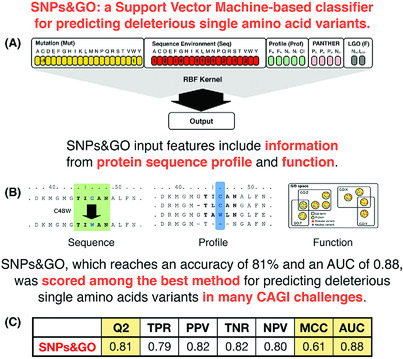
(A) Representation of the SNPs&GO Support Vector Machine-based algorithm. (B) Main input features in SNPs&GO. (C) Performance of SNPs&GO on 38,460 single amino acid variants (Capriotti et al., BMC Genomics, 2013). Evaluation measures (Q2, TPR, PPV, TNR, NPV, MCC and AUC) are defined in Supplementary Materials.
Objective assessment of the evolutionary action equation for the fitness effect of missense mutations across CAGI-blinded contests
- Pages: 1072-1084
- First Published: 23 May 2017
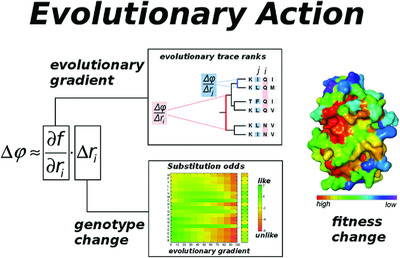
The Evolutionary Action equation is a first order perturbation form of the genotype-phenotype relationship, which predicts the action of genetic variants on fitness. This analytical approach is general and its terms can be numerically estimated from sequence data. Thus, in contrast to the current state-of-the-art methods, it requires no training over large datasets and numerous features. The objective assessments of the Critical Assessment of Genome Interpretation experiments consistently ranked Evolutionary Action one of the most accurate predictors.
PON-P and PON-P2 predictor performance in CAGI challenges: Lessons learned
- Pages: 1085-1091
- First Published: 22 February 2017
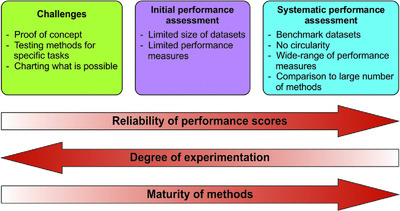
We have participated in various challenges in the four CAGI experiments. We used predictions from PON-P and PON-P2 and additional details to develop models for addressing the challenges. Here, we summarized our approaches and their performances in challenge assessments. We discussed the types of lessons we earned from CAGI, impacts of CAGI on our research and development of predictor with improved performance. We also make some suggestions for future experiments.
Missense variant pathogenicity predictors generalize well across a range of function-specific prediction challenges
- Pages: 1092-1108
- First Published: 16 May 2017
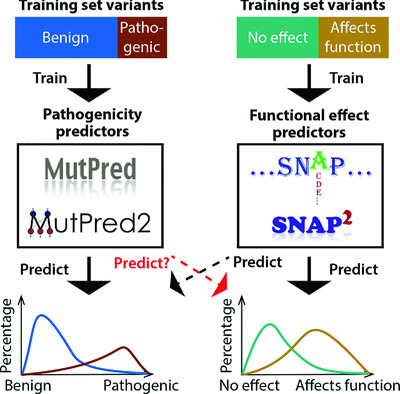
By participating in the Critical Assessment of Genome Interpretation, we demonstrate the direct applicability of missense variant pathogenicity predictors in the task of the prediction of real-valued impact on biochemical, molecular and cellular function, as measured in in vitro experiments. Our work suggests that when a large number of structural and functional features are integrated into a learning algorithm that outputs smooth score distributions, pathogenicity predictors can model the biology shared by both of these prediction tasks.
Ensemble variant interpretation methods to predict enzyme activity and assign pathogenicity in the CAGI4 NAGLU (Human N-acetyl-glucosaminidase) and UBE2I (Human SUMO-ligase) challenges
- Pages: 1109-1122
- First Published: 24 May 2017
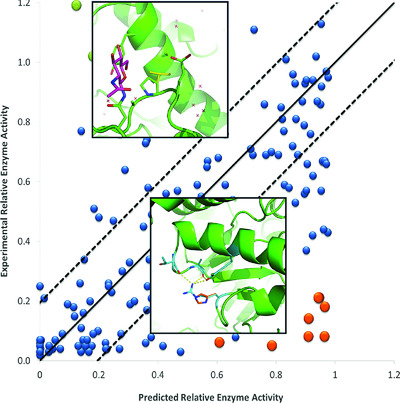
We report results obtained using newly-developed ensemble methods to address two CAGI4 challenges requiring prediction of continuous protein activity: population rare variants in NAGLU that are representative of clinical encounters, and mutations from high throughput mutational scans on UBE2I. The methods are effective, ranking 2nd for SUMO-ligase and 3rd for NAGLU. The ensemble approaches also show considerable potential for estimating the reliability of pathogenic assignments, and are already reliable enough for use with a subset of mutations.
Benchmarking predictions of allostery in liver pyruvate kinase in CAGI4
- Pages: 1123-1131
- First Published: 29 March 2017
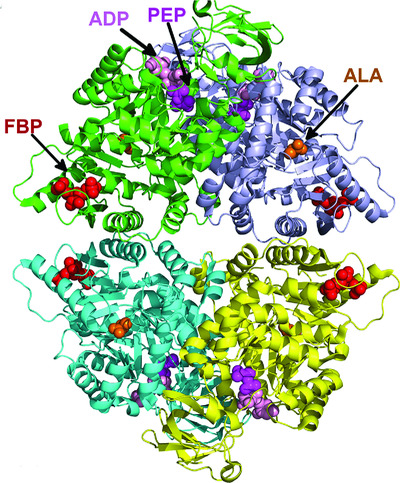
Homotetramer of human liver pyruvate kinase (PYK) with bound substrate (phosphoenol pyruvate), inhibiting allosteric effector (alanine), and activating allosteric effector (fructose-1,6-bisphosphate), compiled from multiple crystal structures of PYK.
Whole-protein alanine-scanning mutagenesis of allostery: A large percentage of a protein can contribute to mechanism
- Pages: 1132-1143
- First Published: 13 April 2017
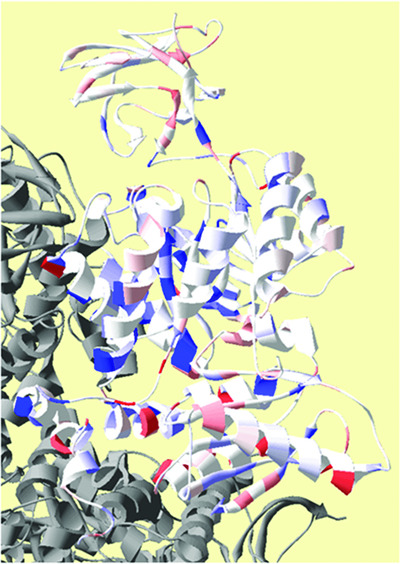
Residue positions at which side chain removal alters allosteric coupling for Fru-1,6-BP activation (Qax-Fru-1,6-BP).
Exploring the limits of the usefulness of mutagenesis in studies of allosteric mechanisms
- Pages: 1144-1154
- First Published: 29 April 2017
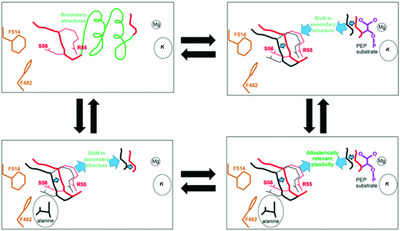
A simplistic allosteric mechanism model based on allosterically-relevant plasticity. This model was developed with acknowledged insufficient structural insights, but provided a framework for mutational probings of the allosteric binding site. Although we provide data that is not consistent with this model, the responses of many mutant proteins could be viewed as consistent, raising questions about how mutational probings should be used in studies of allosteric mechanisms.
Lessons from the CAGI-4 Hopkins clinical panel challenge
- Pages: 1155-1168
- First Published: 11 April 2017
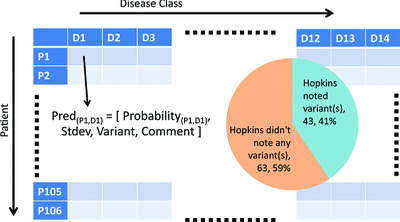
The CAGI-4 Hopkins clinical panel challenge was an attempt to assess state-of-the-art methods for clinical phenotype prediction from DNA sequence. Participants were provided with exonic sequences of 83 genes for 106 patients from the Johns Hopkins DNA Diagnostic Laboratory. Five groups participated in the challenge, predicting both the probability that each patient had each of the 14 possible classes of disease, as well as causal variants. We discuss the accuracy of the predictions and their implications for further methods development.
CAGI4 SickKids clinical genomes challenge: A pipeline for identifying pathogenic variants
- Pages: 1169-1181
- First Published: 16 May 2017
The CAGI4 SickKids clinical genome challenge provided an opportunity to assess the landscape of genetic variants found in a difficult set of 25 unsolved rare disease cases. We used a three-stage pipeline consisting of careful analysis of data quality, classification of disease relevant gene-specific variants into seven categories, and prioritization of variants by compatibility with the complex phenotype of a patient.We were able to determine the genetic cause of a case of epileptic encephalopathy, a missense mutation in KCNB1.
Working toward precision medicine: Predicting phenotypes from exomes in the Critical Assessment of Genome Interpretation (CAGI) challenges
- Pages: 1182-1192
- First Published: 21 June 2017
The Critical Assessment of Genome Interpretation (CAGI) is a community experiment consisting of genotype-phenotype prediction challenges; participants build models, undergo assessment, and share key findings. For CAGI 4, three challenges involved using exome sequencing data: Crohn's disease, bipolar disorder, and warfarin dosing. We discuss the range of techniques used for phenotype prediction, the methods used for assessing predictive models, and the lessons gleaned from the CAGI exomes challenges.
Crohn disease risk prediction—Best practices and pitfalls with exome data
- Pages: 1193-1200
- First Published: 13 January 2017
Determination of disease phenotypes and pathogenic variants from exome sequence data in the CAGI 4 gene panel challenge
- Pages: 1201-1216
- First Published: 12 May 2017
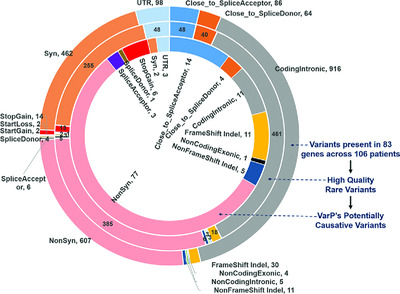
We describe the design and implementation of a gene panel sequencing data analysis pipeline, VarP. The performance of the pipeline was assessed in the CAGI 4 community experiment. VarP identified the correct disease class and potentially causative variant(s) in 36/106 patients, including 10 patients where the clinical pipeline did not find any causative variants. Post analysis showed that use of three-dimensional structure could have assisted interpretation in a number of cases.
DeepBipolar: Identifying genomic mutations for bipolar disorder via deep learning
- Pages: 1217-1224
- First Published: 10 June 2017

We designed an end-to-end deep learning architecture (called DeepBipolar) to predict bipolar disorder based on limited genomic data. Leveraging Deep Convolutional Neural Network (DCNN), it automatically extracts features from genotype information to predict the bipolar phenotype. DeepBipolar was considered the most successful by the independent assessor in the Critical Assessment of Genome Interpretation (CAGI) bipolar disorder challenge.
CAGI4 Crohn's exome challenge: Marker SNP versus exome variant models for assigning risk of Crohn disease
- Pages: 1225-1234
- First Published: 16 May 2017
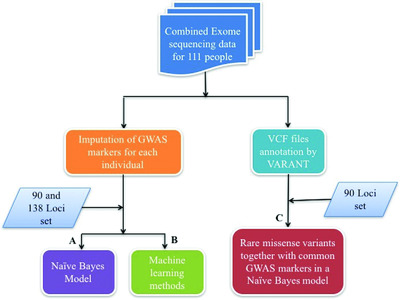
Understanding the basis of complex trait disease is a fundamental problem in human genetics. The three rounds of CAGI Crohn's Exome challenges provide insight into the adequacy of current disease models by requiring participants to identify which of a set of individuals has been diagnosed with the disease, given exome data. For the CAGI4 round, we developed a method that uses machine learning methods to assign disease risk, with only the imputed genotypes of GWAS marker SNPs as input.
Stratified polygenic risk prediction model with application to CAGI bipolar disorder sequencing data
- Pages: 1235-1239
- First Published: 17 April 2017
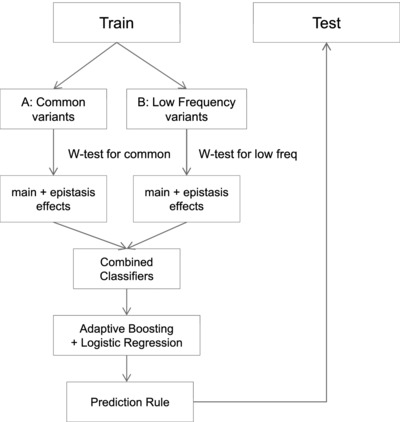
This study proposed to perform complex trait prediction using a stratified design. The genetic data are divided into strata according to genetic architectures, and feature selection is conducted within each strata through a data adaptive W-test for main effect and pairwise interactions. An ensemble classification algorithm can be applied to integrate the selected features to perform prediction. Application on CAGI data set showed that including interaction effect of common variants improved prediction accuracy.
Predicting gene expression in massively parallel reporter assays: A comparative study
- Pages: 1240-1250
- First Published: 21 February 2017
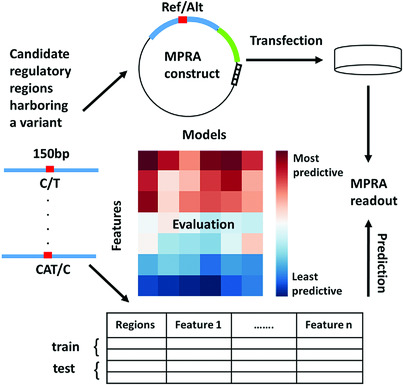
Deciphering the functionality of the non-coding genome has been the focus of many recent studies, aiming to annotate regulatory regions and understand their specific role in disease and other phenotypes. The goal of the CAGI eQTL challenge was to predict the activity of candidate genomic regions (experimentally evaluated by massively parallel reporter assays). Our meta-analysis of competing submissions highlights features and models that lead to accurate prediction and points to areas for improvement.
Predicting enhancer activity and variant impact using gkm-SVM
- Pages: 1251-1258
- First Published: 25 January 2017

DNA variation in regulatory regions (e.g. enhancers) has been implicated as contributing to many human diseases. We have been developing quantitative computational models of regulatory regions to predict the impact of this variation and to identify the mechanisms of enhancer modulation. In this blind test, our model predictions are in quantitative agreement with direct measurements of enhancer activity.
Accurate eQTL prioritization with an ensemble-based framework
- Pages: 1259-1265
- First Published: 21 February 2017
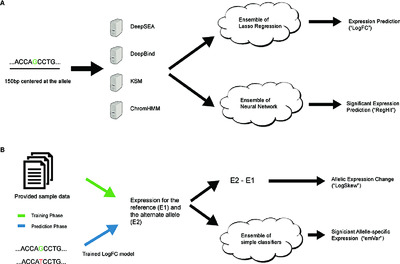
We present a novel ensemble-based computational framework, EnsembleExpr, that achieved the best performance in the Fourth Critical Assessment of Genome Interpretation expression quantitative trait locus “(eQTL)-causal SNPs” challenge for identifying eQTLs and prioritizing their gene expression effects. We envision EnsembleExpr to be a resource to help interpret noncoding regulatory variants and prioritize disease-associated mutations for downstream validation.
Matching phenotypes to whole genomes: Lessons learned from four iterations of the personal genome project community challenges
- Pages: 1266-1276
- First Published: 23 May 2017
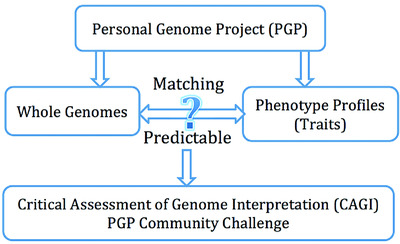
PGP provides unrestricted access to genomes of individuals and their associated phenotypes. The CAGI PGP challenge is to predict whether an individual had a particular trait or phenotype profile based on their whole genome. Assessment results show prediction of individual traits is difficult, relying on a strong knowledge of trait frequency within general population, while matching genomes to trait profiles relies heavily upon a small number of common traits.