

Journal list menu

 Issue2014i-v, 1-150
Issue2014i-v, 1-150
Export Citations
Download PDFs
When to WES in the Pediatric Disease Clinic? Now!
- Page: v
- First Published: 18 December 2013
A Role for MSH2 in the CGG Repeat Expansion
- Page: v
- First Published: 18 December 2013
Mitochondrial DNA Rearrangements in Health and Disease—A Comprehensive Study
- Pages: 1-14
- First Published: 01 October 2013
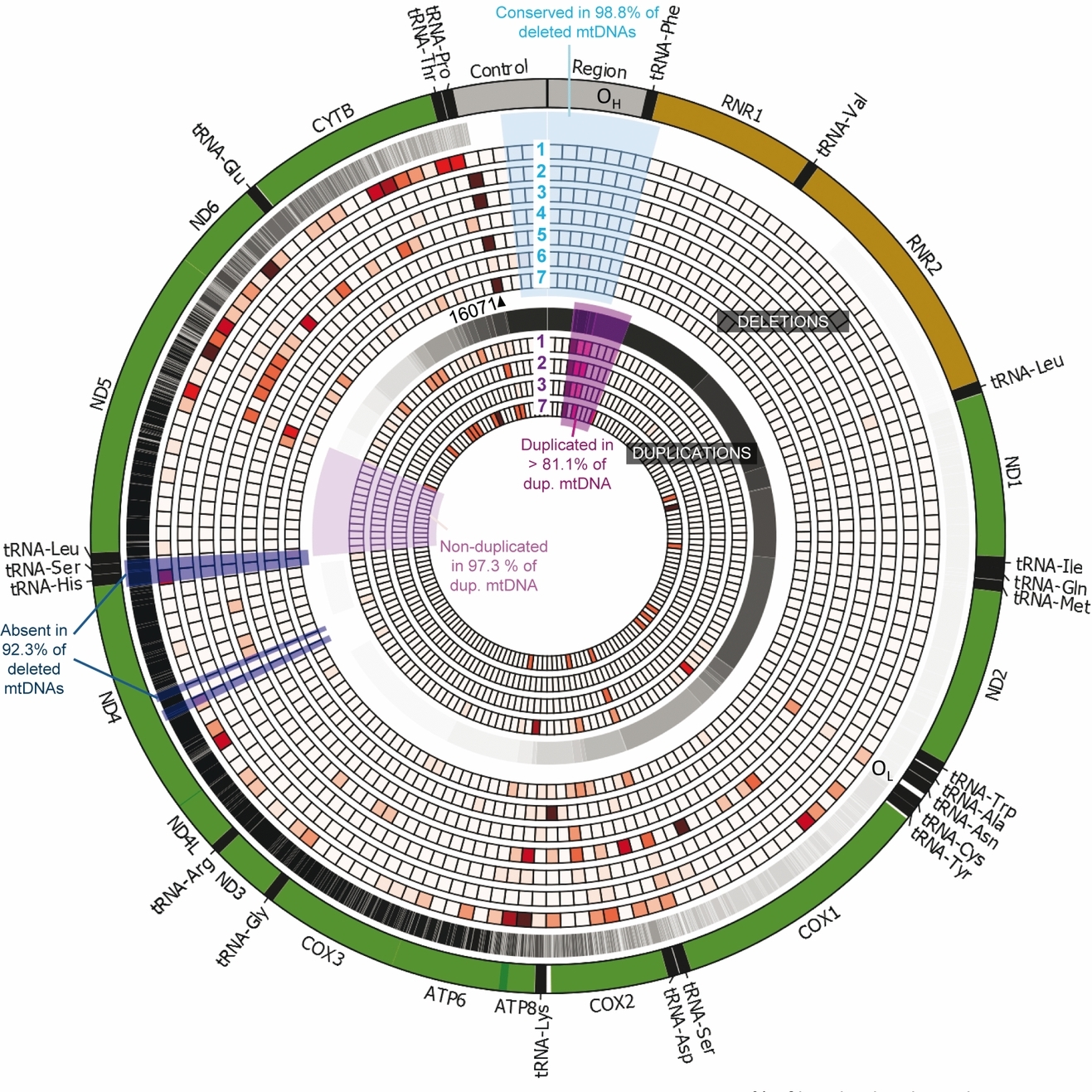
Mitochondrial DNA (mtDNA) rearrangements are a primary cause of mitochondrial disease and have been implicated in aging and various neurodegenerative disorders. Here we present a comprehensive study of more than 750 mtDNA deletions and duplications described over the last 30 years. Our study reveals striking differences among various diseases in the distribution of mtDNA rearrangements and provides a detailed catalogue of rearranged mtDNA molecules to guide future investigations.
Genetic Basis of Congenital Erythrocytosis: Mutation Update and Online Databases
- Pages: 15-26
- First Published: 30 September 2013
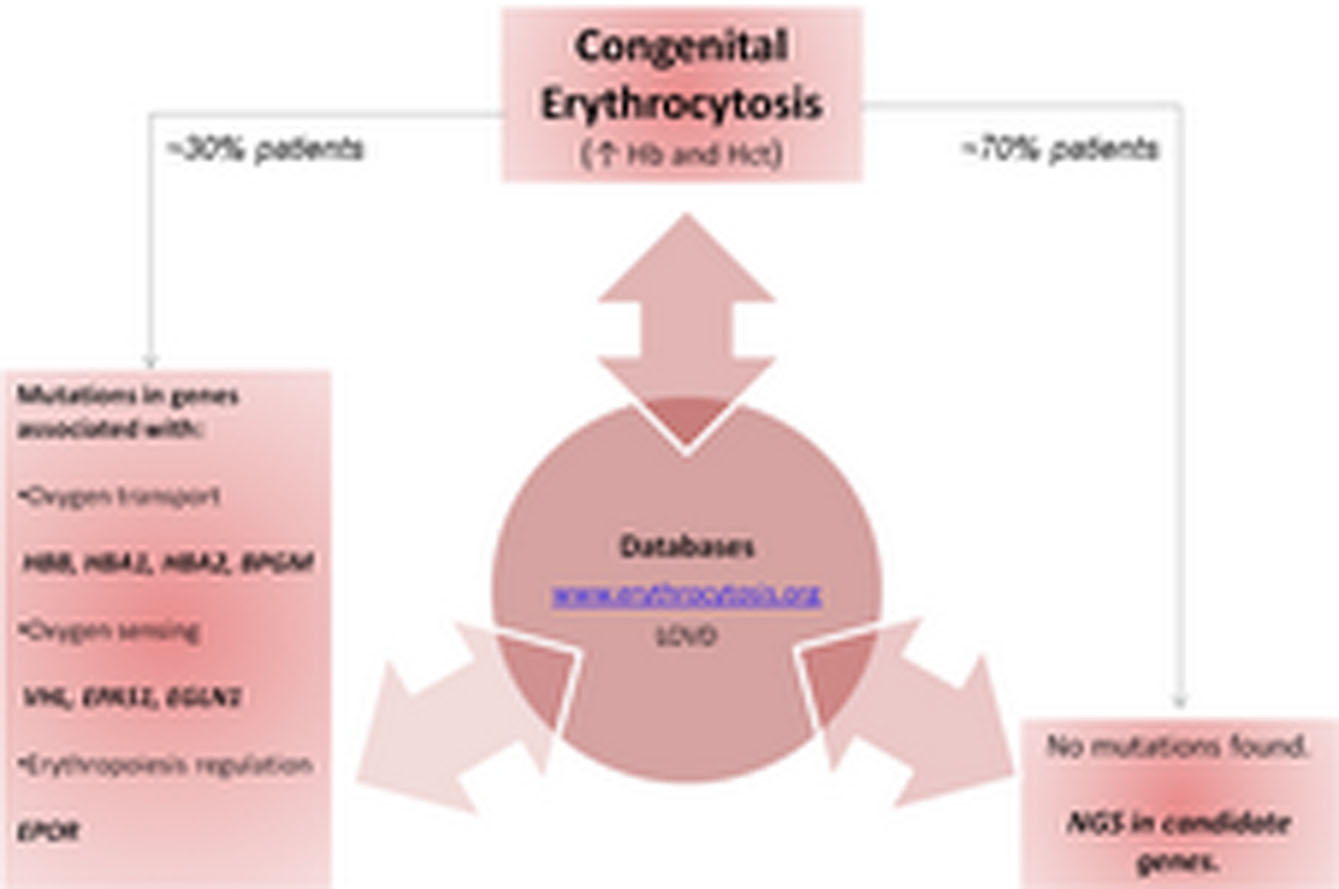
Congenital Erythrocytosis (CE) is caused by deregulated red blood cell production where erythrocyte overproduction results in elevated hemoglobin and hematocrit levels. Despite recent important discoveries in the molecular pathogenesis of CE, the molecular causes remain to be identified in about 70% of the patients. The implementation of internet-based erythrocytosis database and next-generation sequencing is expected to further expand the number of genes involved in CE and to establish clinical and genotype–phenotype correlations in larger groups of individuals.
Mutations and Polymorphisms in the Human Argininosuccinate Lyase (ASL) Gene
- Pages: 27-35
- First Published: 25 October 2013
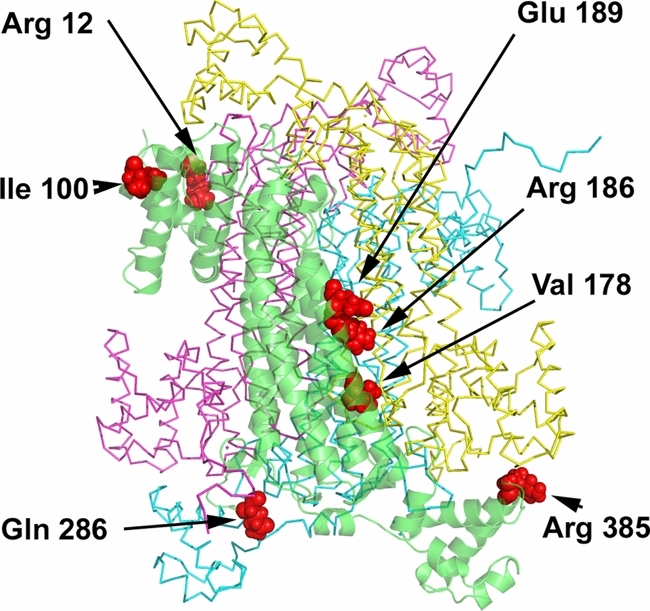
Argininosuccinate lyase deficiency (ASLD) is an inborn error of metabolism caused by a defect of the urea cycle enzyme argininosuccinate lyase (ASL) encoded by the ASL gene. Patients are affected by life-threatening hyperammonemia, mental retardation and liver disease. We are providing here an update on the molecular basis of this disease by presenting all today known mutations (n = 134) as well as all known different genotypes (n = 160). To illustrate these data, we are presenting structural considerations focussing on the relevance of the known mutations for ASL homotetramer formation and function.
Ciliary Genes TBC1D32/C6orf170 and SCLT1 are Mutated in Patients with OFD Type IX
- Pages: 36-40
- First Published: 06 November 2013
Alkuraya and colleagues identify truncating mutations in two ciliary proteins in patients with OFD XI.
Targeted Deep Resequencing Identifies MID2 Mutation for X-Linked Intellectual Disability with Varied Disease Severity in a Large Kindred from India
- Pages: 41-44
- First Published: 02 October 2013
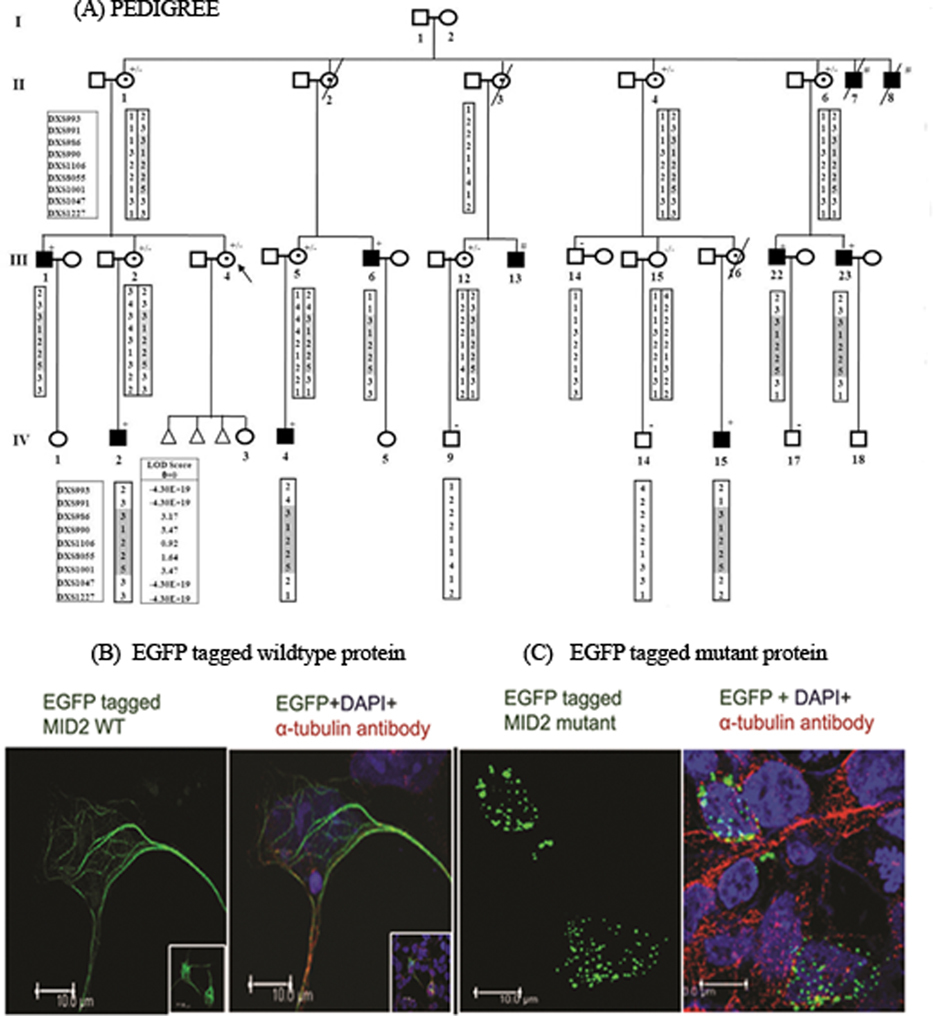
A novel missense mutation in MID2 was identified in a large kindred with varying degree of X-linked intellectual disability, on targeted resequencing of a linked region. When transiently expressed in HEK293T cell line, the mutation abolished the function of the COS domain of the protein, resulting in altered sub-cellular localization. This study highlights the growing role of the ubiquitin pathway in intellectual disability and also the difference in disease phenotype compared with that caused by mutations in its paralogue MID1.
Exome Sequencing as a Diagnostic Tool for Pediatric-Onset Ataxia
- Pages: 45-49
- First Published: 01 October 2013
GALNT12 is Not a Major Contributor of Familial Colorectal Cancer Type X
- Pages: 50-52
- First Published: 01 October 2013
Capillary Electrophoresis Analysis of Conventional Splicing Assays: IARC Analytical and Clinical Classification of 31 BRCA2 Genetic Variants
- Pages: 53-57
- First Published: 07 October 2013
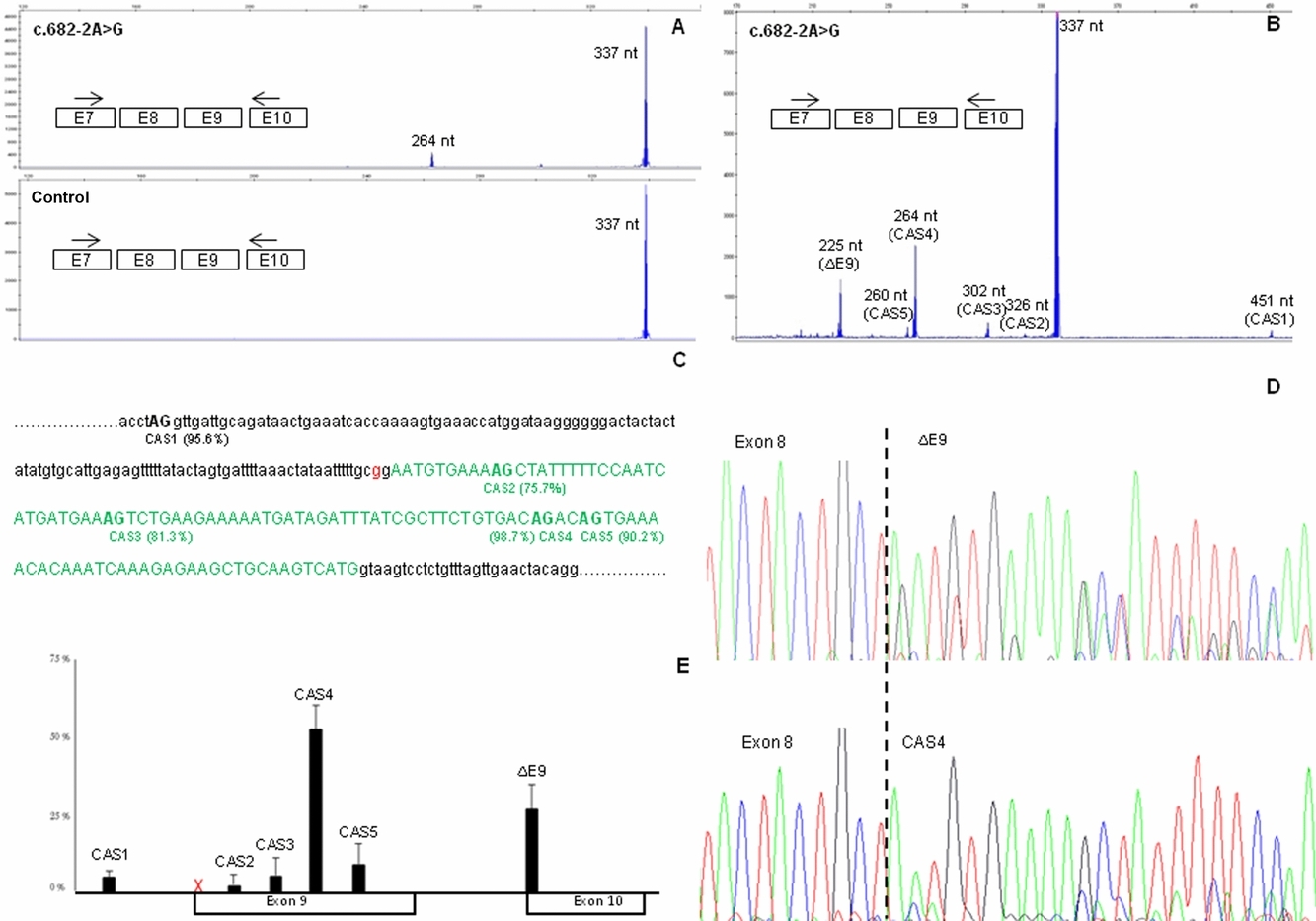
Splicing analyses are widely used in the clinical setting to address the significance of rare genetic variants. However, interpretation of results is often challenging. Here we show that incorporating capillary electrophoresis (EP) and direct sequencing to the analysis of RT-PCR splicing assays often produces clear-cut analytical outputs. Indeed, we have produced clear-cut outputs in 29 out of 31 BRCA2 variants investigated, including the identified of clinically relevant splicing aberrations, and one splicing quantitative trait locus (sQTL).
Variant ATRX Syndrome with Dysfunction of ATRX and MAGT1 Genes
- Pages: 58-62
- First Published: 15 October 2013
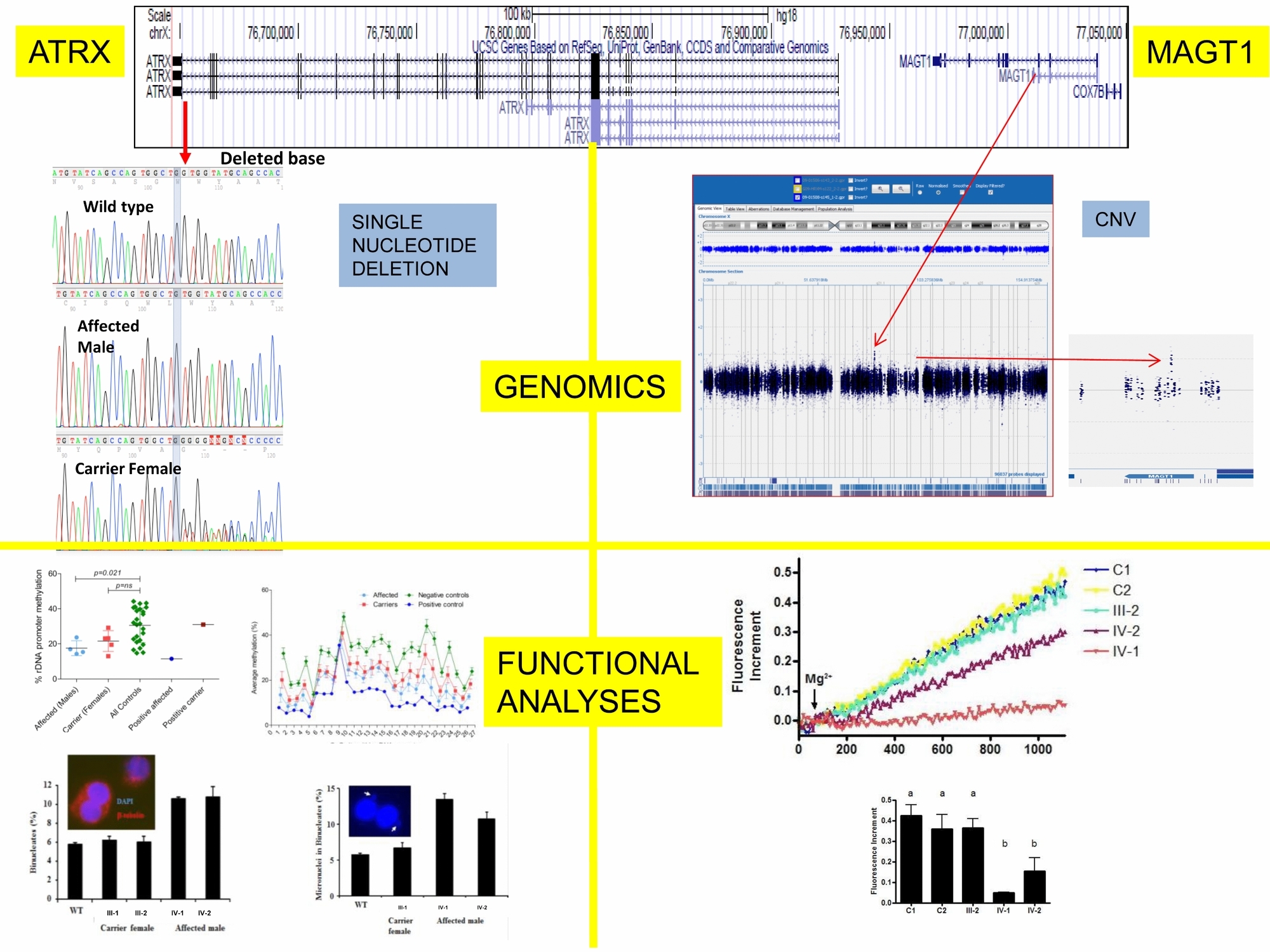
A 0.8kb intronic duplication in MAGT1 and a single base pair deletion in the last exon of ATRX were identified in a family with 5 males demonstrating intellectual disability (ID) and unusual skin findings. Dysfunction of MAGT1 and ATRX was detected using a panel of tests and indicates the role of ATRX mutation in ID and MAGT1 disruption in abnormal skin findings. This work emphasizes the importance of correlating clinical phenotype with genomic and cell function analyses to explain the phenotypic variability of syndromic ID.
High Frequency Strand Slippage Mutations in CTCF in MSI-Positive Endometrial Cancers
- Pages: 63-65
- First Published: 15 October 2013
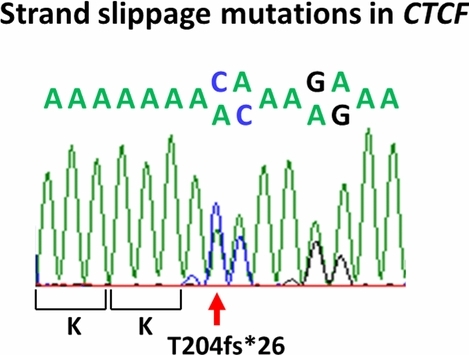
The chromatin binding factor CTCF was recently shown to be as a significantly mutated gene in endometrial cancers. We identified strand slippage mutations in CTCF in > 25% of tumors with defective DNA mismatch repair. The previously unreported insertion and deletion mutations in an A7 coding repeat are subject to nonsense mediated decay, and based on the observed mutations we propose CTCF is a haploinsufficient tumor suppressor.
Genetic and Functional Analyses of ZIC3 Variants in Congenital Heart Disease
- Pages: 66-75
- First Published: 07 October 2013
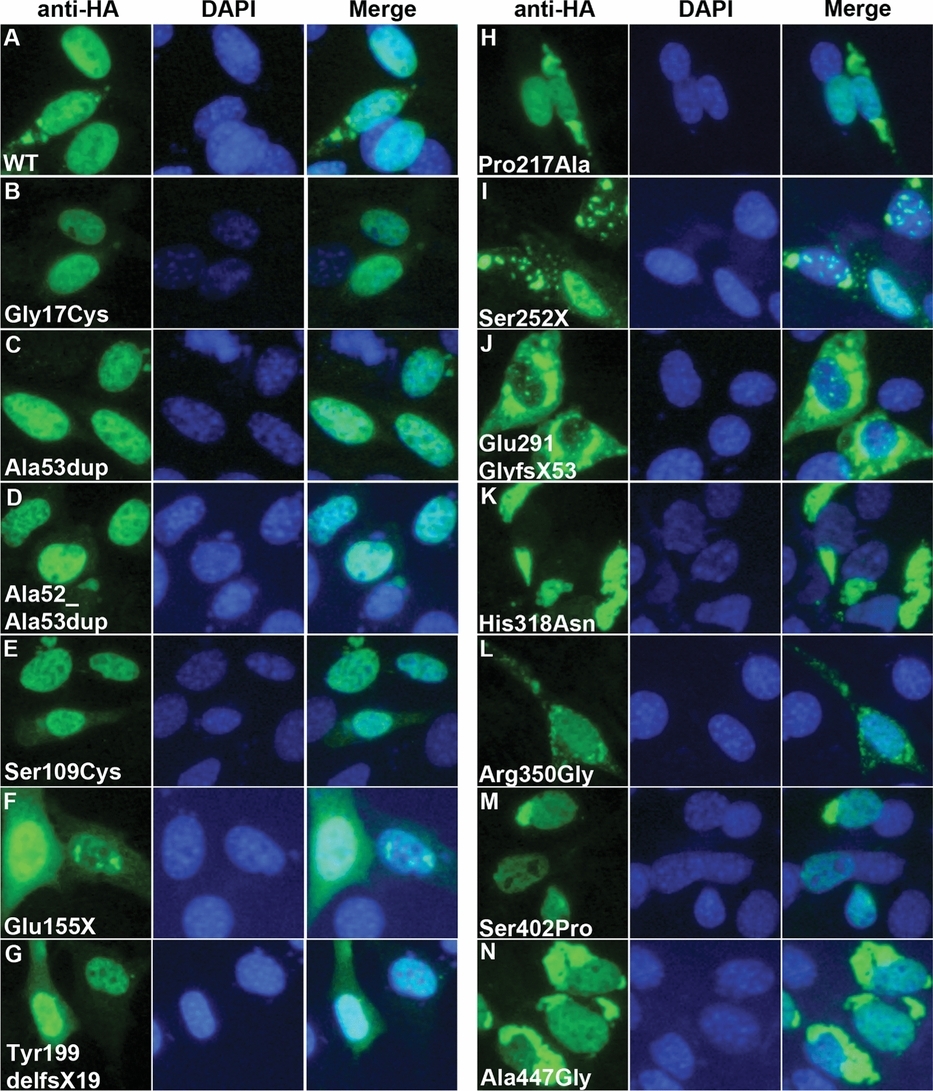
Mutations in ZIC3 result in heterotaxy, a condition characterized by abnormal organ laterality, complex cardiovascular malformations, and isolated congenital heart disease (CHD). We identified 11 mutations (eight novel) in 440 unrelated patients with heterotaxy and CHD. Aberrant cytoplasmic localization and disrupted nuclear function was observed for zinc-finger mutations, but not for mutations in other regions. Our data expands the ZIC3 mutational spectrum, supports a higher than expected prevalence in sporadic cases (3.8%), and suggests alternative functions for terminal ZIC3 mutations.
Extreme Growth Failure is a Common Presentation of Ligase IV Deficiency
- Pages: 76-85
- First Published: 10 October 2013
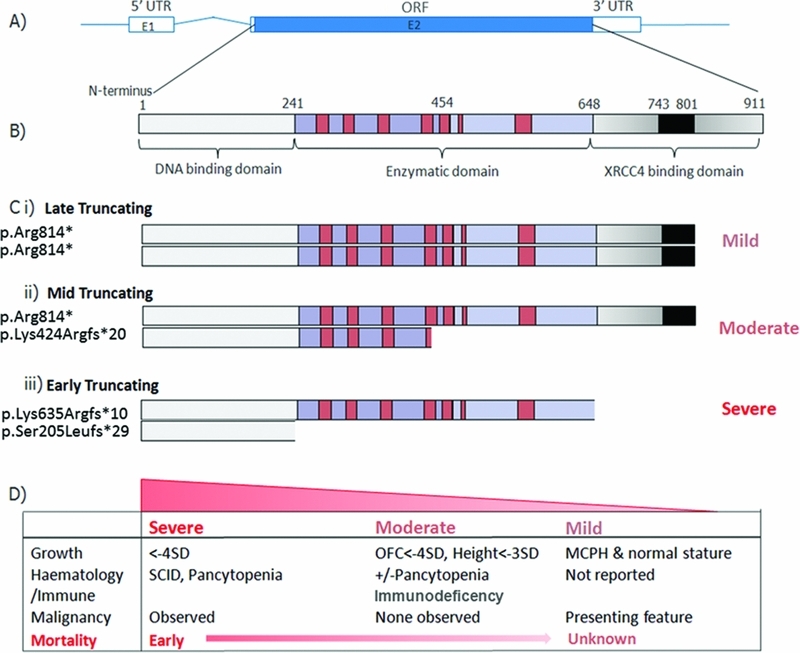
Ligase IV syndrome, a rare disorder of DNA damage repair, is associated with severe immunodeficiency, malignancy predisposition, and microcephaly. Here, we report that mutations in LIG4 also occur in patients presenting with extreme growth failure, extending the phenotypic spectrum of Ligase IV deficiency. A correlation between degree of protein truncation and disease severity is observed, with a recurrent mutation presenting in conjunction with a more severely truncating mutation conferring profound growth impairment with progressive bone marrow failure and immunodeficiency.
The ETFDH c.158A>G Variation Disrupts the Balanced Interplay of ESE- and ESS-Binding Proteins thereby Causing Missplicing and Multiple Acyl-CoA Dehydrogenation Deficiency
- Pages: 86-95
- First Published: 07 October 2013
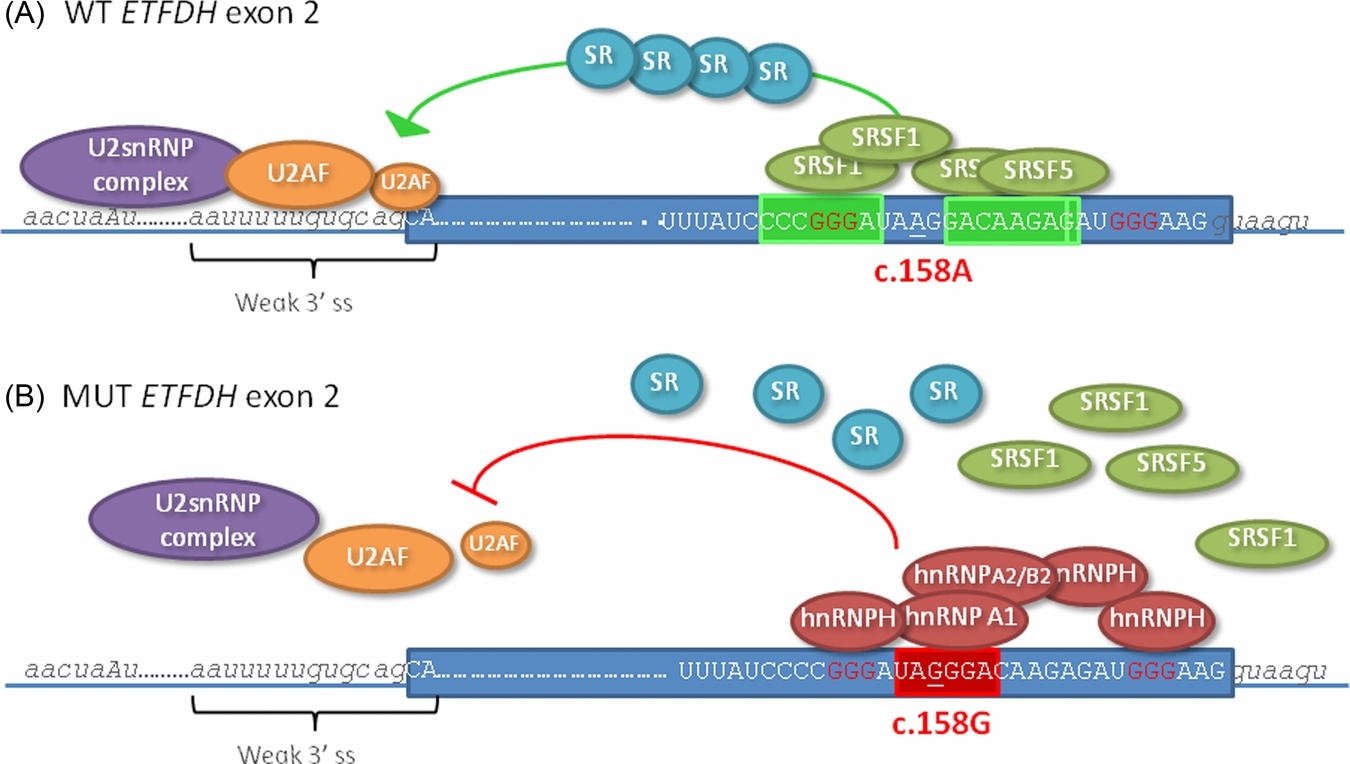
It is demonstrated that a predicted benign ETFDH missense variation instead causes exon 2 skipping and disease. This mutation creates a general splicing inhibitory element, which also cause aberrant splicing in other disease genes. The element binds several splicing inhibitory proteins, such as hnRNPA1, hnRNPA2 and hnRNPH. We hypothesize that the splicing inhibitory effect of this element in ETFDH is further enhanced by its location in a sequence context with flanking hnRNPH-binding GGG triplets, which may act synergistically to inhibit splicing.
An NTD-Associated Polymorphism in the 3′ UTR of MTHFD1L can Affect Disease Risk by Altering miRNA Binding
- Pages: 96-104
- First Published: 10 October 2013
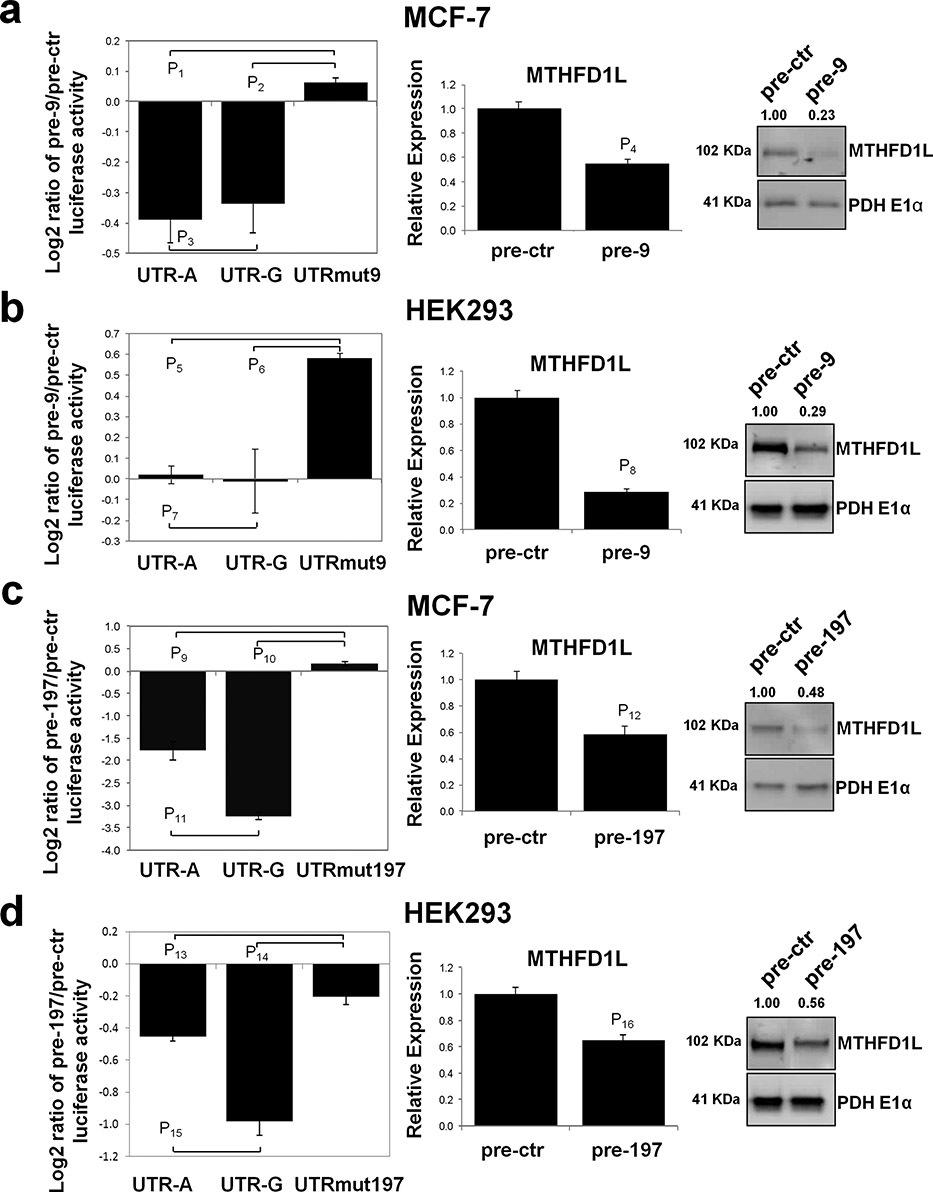
Altered expression levels of MTHFD1L, a gene involved in folate metabolism, have an impact on a number of common diseases including neural tube defects (NTDs) and cancer. This article demonstrates that MTHFD1L is regulated by the microRNA miR-197, and its binding efficacy is affected by the single nucleotide polymorphism (SNP) rs7646 in the 3' Untranslated Region. SNP rs7646 has previously been associated with maternal risk of NTDs.
Exome Sequencing Identifies Potential Risk Variants for Mendelian Disorders at High Prevalence in Qatar
- Pages: 105-116
- First Published: 10 October 2013
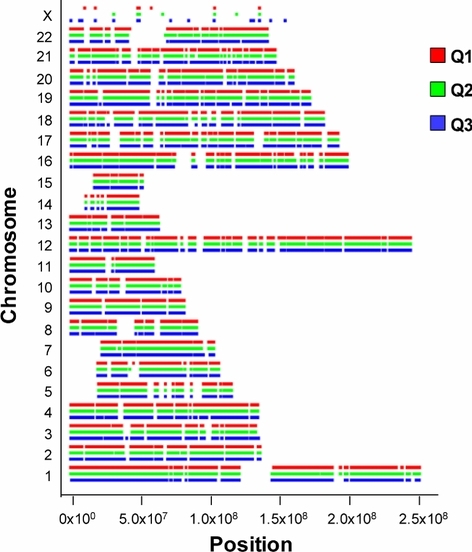
This study presents a framework for enabling precision genome-based medicine (PGM) in a population not sampled by nor related to public consortium sequencing projects, using the example of Qatar. Through sampling and exome sequencing of representatives from three major ancestry groups identified in prior work (Q1 Bedouin, Q2 Persian-South Asian, Q3 African), we identified 37 variants in 33 genes with effects on 36 clinically significant Mendelian diseases. Genetic screening in Qatar includes only 4 out of the 37. This study provides a set of Mendelian disease variants with potential impact on the epidemiological profile of the population that could be incorporated into the testing program if further experimental and clinical characterization confirms high penetrance.
Correlation of Phenotype/Genotype in a Cohort of 23 Xeroderma Pigmentosum-Variant Patients Reveals 12 New Disease-Causing POLH Mutations
- Pages: 117-128
- First Published: 15 October 2013
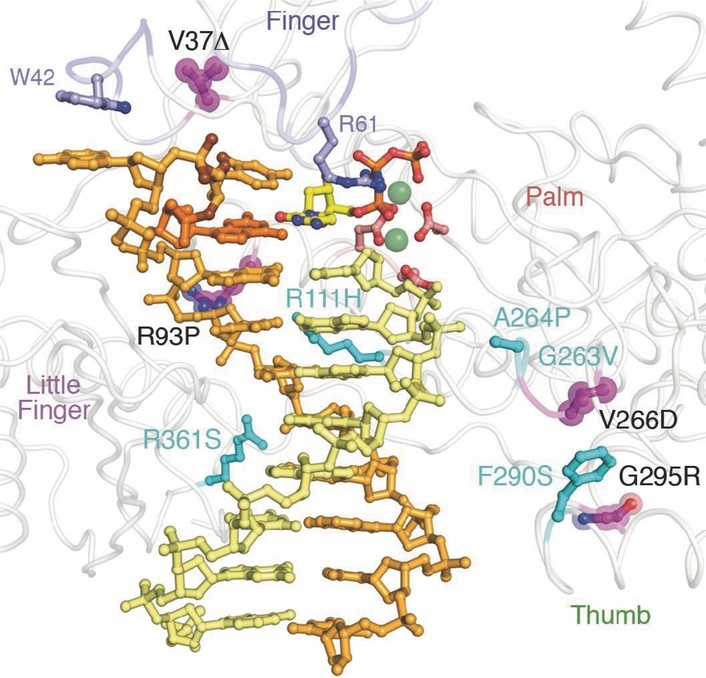
Point mutations located in the active site of the translesion polymerase η dictate the clinical aggressiveness of the xeroderma pigmentosum variant syndrome. The mutated proteins are unable to faithfully replicate through UV-induced DNA lesions leading, therefore, to numerous skin cancers. This picture represents a replication fork with orange parental template and yellow primer strand. The mutations in the active domain are highlighted (for example R93P: Arg93Pro) in different colors.
The Mismatch Repair Protein MSH2 is Rate Limiting for Repeat Expansion in a Fragile X Premutation Mouse Model
- Pages: 129-136
- First Published: 15 October 2013
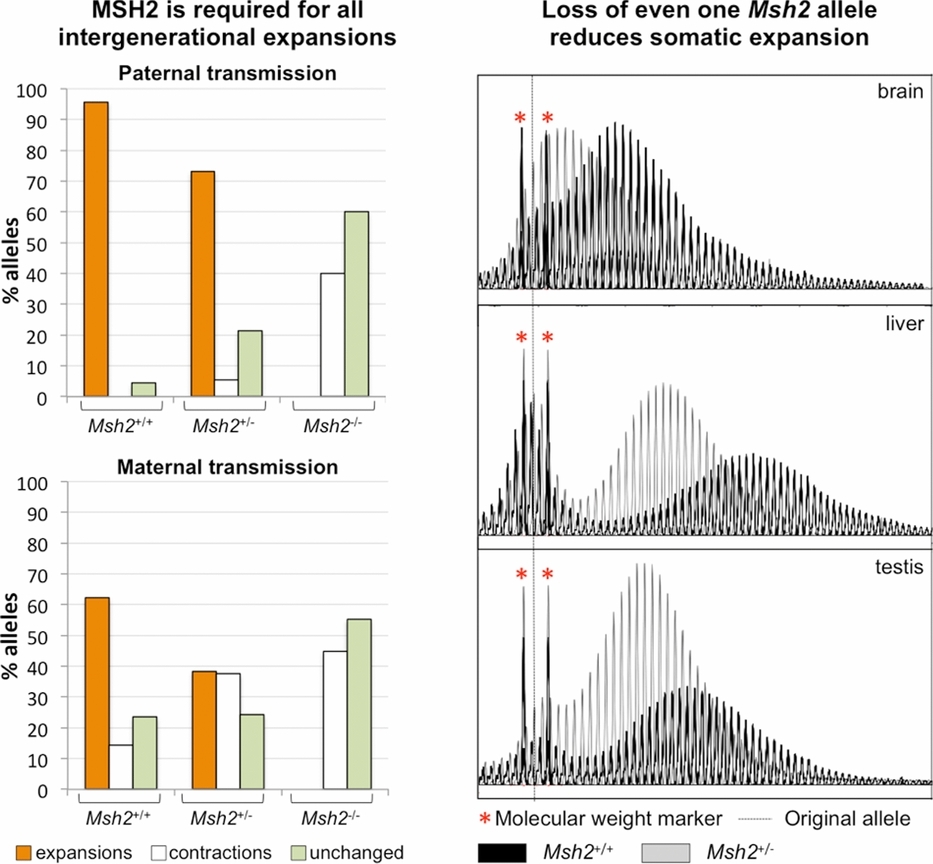
The Fragile X-related disorders result from expansion of a CGG/CCG-repeat tract (microsatellite) at the 5' end of the FMR1 gene. We show here that the mismatch repair protein MSH2 is required for all germ-line and somatic expansion mutations in a Fragile X mouse model. MSH2 is best known for its role in reducing the microsatellite instability that would otherwise be manifest in certain kinds of cancer. How MSH2 protects against one form of microsatellite instability yet causes another is unknown
A Homozygous PDE6D Mutation in Joubert Syndrome Impairs Targeting of Farnesylated INPP5E Protein to the Primary Cilium
- Pages: 137-146
- First Published: 25 October 2013
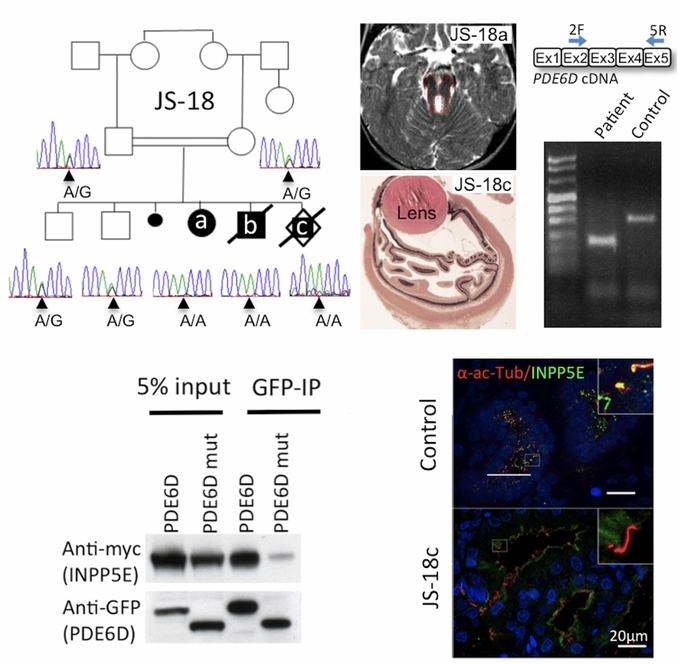
In a consanguineous family with Joubert syndrome, we identified a homozygous splice site mutation in PDE6D, encoding a prenyl-binding protein. Proteomic analysis identified INPP5E, whose mutations also lead to JS or MORM syndromes, as novel prenyl-dependent cargo of PDE6D. Mutant PDE6D shows reduced binding to INPP5E, which fails to localize to primary cilia indicating the requirement of functional PDE6D for correct targeting of INPP5E to the primary cilia. This study provides the first evidence of prenylbinding-dependent trafficking in ciliopathies.
Comprehensive Registration of DNA Sequence Variants Associated with Inherited Retinal Diseases in Leiden Open Variation Databases
- Pages: 147-148
- First Published: 10 October 2013
Analysis of LMNB1 Duplications in Autosomal Dominant Leukodystrophy Provides Insights into Duplication Mechanisms and Allele-Specific Expression
- Page: 149
- First Published: 28 October 2013
Screening of a Large Cohort of Leber Congenital Amaurosis and Retinitis Pigmentosa Patients Identifies Novel LCA5 Mutations and New Genotype–Phenotype Correlations
- Page: 150
- First Published: 30 October 2013