

High Frequency Strand Slippage Mutations in CTCF in MSI-Positive Endometrial Cancers
Communicated by Mark H. Paalman
Contract grant sponsor: National Institutes of Health (R21 CA155674, U01 HG006517).
ABSTRACT
Tumors with defective mismatch repair acquire large numbers of strand slippage mutations including frameshifts in coding sequence repeats. We identified a mutational hotspot, p.T204fs, in the insulator-binding protein (CTCF) in MSI-positive endometrial cancers. Although CTCF was described as a significantly mutated gene by the endometrial cancer TCGA, the A7 track variants leading to T204 frameshifts were not reported. Reanalysis of TCGA data using Pindel revealed frequent T204fs mutations, confirming CTCF is an MSI target gene and revealed the same frameshifts in tumors with intact mismatch repair. We show that T204fs transcripts are subject to nonsense-mediated decay and as such, T204fs mutations are unlikely to act as dominant negatives. The spectrum and pattern of mutations observed is consistent with CTCF acting as a haploinsufficient tumor suppressor.
Endometrial cancer is the most common gynecologic malignancy in the United States. The majority of endometrial cancers are histologically classified as endometrioid adenocarcinomas [Siegel et al., 2012]. Endometrioid tumors frequently have defective DNA mismatch repair that leads to tumor microsatellite instability (MSI) [Duggan et al., 1994]. MSI is associated with elevated mutation frequency and in particular, strand slippage mutations (frameshifts) in simple repeats. Coding sequence repeats in a number of loci have been reported (MSH3, BAX, ATR, TARBP2, and others) [Melo et al., 2009; Schwartz et al., 1999; Swisher et al., 1998; Vassileva et al., 2002; Zighelboim et al., 2009]. However, unlike colon cancers with MSI, for which several MSI target genes have been described (TGFRB2, BAX, and additional loci [Markowitz et al., 1995]), bona fide MSI mutational targets in endometrial cancer remain elusive. We identified frequent insertion/deletion mutations in CCCTC-binding insulator and transcription factor (CTCF; MIM #604167) in MSI-positive endometrial cancers.
We performed exome sequencing for primary tumors from eight women with early stage MSI-positive endometrioid adenocarcinomas who subsequently recurred (Supp. Methods). Three tumors carried CTCF variants: one with a p.R29W substitution and two with an insertion, p.T204fs (Fig. 1A). Neither variant was reported by the endometrial Cancer Genome Atlas (TCGA) [Kandoth et al., 2013].

Analysis of the CTCF coding region in an additional 149 primary endometrioid tumors (98 MSI-positive and 51 with normal mismatch repair previously assessed for MSI as described [Kandoth et al., 2013; Zighelboim et al., 2009]) proved that the A7 track in exon 3 is a mutational hot spot in cancers with defective DNA mismatch repair: 25% of MSI-positive cases carried either the A insertion mutation initially identified in our exome sequencing studies, or an A deletion mutation (17 cases with p.T204fs*26 and 8 cases with p.T204fs*18, respectively) (Table 1). The overall mutation rate was 35% for MSI-positive tumors and 20% for MSI-negative tumors, with two additional MSI-positive tumors having silent mutations. The vast majority of somatic variants identified are almost certainly loss of function mutations (32 frameshifts, five stops, and three splice site mutations) with only six missense mutations seen (four in MSI-positive tumor and two in MSI-negative tumors). It is noteworthy that among the 43 cases with mutations only two had a second mutation: one microsatellite stable tumor carried two frameshift mutations (p.V50fs and p.M285fs) and an MSI-positive tumor had a stop and two missense mutations (p.R457*, p.A694V, and p.L722I) (Table 1). CTCF mutations were more common in low grade tumors than high grade tumors (37% mutation in grade 1, 28% in grade 2, and 19% in grade 3 cancers) but the difference did not reach statistical significance (P = 0.052 for trend). There was no association with FIGO stage, patient BMI or age at diagnosis (Supp. Table S1).
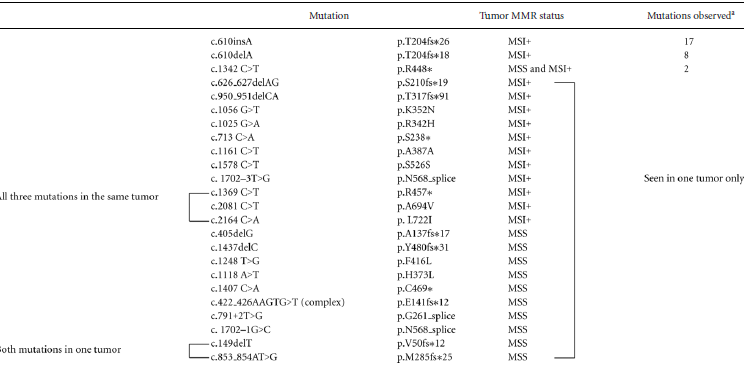 |
- ap.R448*, p.G216_splice c.791+2T>G, and p.N568_splice c.1702–1G>C were reported for endometrial cancer TCGA series (Kandoth et al., 2013).
- Two additional silent mutations identified in two MSI+ tumors (p.S526S, c.1578 C>T and p.T518T, c.1554 C>T).
- MSS: microsatellite stable.
- Note: Mutations listed here for 149 primary tumors do not include p.R29W and two instances of p.T204fs*26 mutations identified in initial exome sequencing study.
- Mutations are referenced to NM_006565.3.
- Nucleotide numbering reflects cDNA numbering with +1 corresponding to the A of the ATG translation initiation codon in the reference sequence (NM_006565.3). The initiation codon is codon 1.
- Variants have been submitted to COSMIC (cancer.sanger.ac.uk/).
The preponderance of frameshift, splice site, and stop mutations seen in endometrial cancers is consistent with CTCF acting as a tumor suppressor. The recurrent frameshift defects (p.T204fs*18 and p.T204fs*26) could act as dominant negatives and thus explain the absence of a “second hit” in cases with those mutations. Reverse transcriptase PCR and sequencing revealed a near complete absence of mutant transcripts in multiple primary tumors with the A insertion or deletion, consistent with A7 insertion/deletion mutation nonsense-mediated decay (Fig. 1B). Primary tumors carrying stop or other frameshift mutations showed similar nonsense-mediated decay of the mutant transcripts, whereas a missense change (p.H373L, c.1118A>T) was present in tumor mRNA.
The absence of mutant transcripts, coupled with the paucity of second mutations in the majority of tumors, suggests haploinsufficiency for CTCF in endometrioid endometrial cancers. PTEN, which is frequently mutated in endometrial cancer, is one example of a haploinsufficient tumor suppressor [Di Cristofano et al., 1998; Joshi et al., 2012]. Given CTCF is an essential gene [Merkenschlager and Odom, 2013] and its critical role in the global regulation of gene expression, it is possible that complete loss of CTCF activity is cell lethal as has been described for DICER1, another haploinsufficient tumor suppressor [Kumar et al., 2009].
CTCF p.T204fs mutations, present in 25% of MSI-positive endometrial cancers in our study, were not reported by the endometrial cancer TCGA [Kandoth et al., 2013]. Analysis of TCGA exome sequences for the endometrioid nonultramutator [Kandoth et al., 2013] cases using Pindel [Ye et al., 2009] did, however, reveal somatic p.T204fs mutations at a frequency comparable to that we observed using Sanger sequencing. Eleven of 63 MSI-high cases (17.5%) had an A track insertion or deletion mutation (seven and four, respectively). The A7 insertion mutation was also seen at low frequency in MSI stable tumors (four of 109, 3.7%) (Supp. Table S2). The fact that the p.T204fs*26 mutation is also seen in microsatellite stable tumors further supports the notion that the A track insertions and deletions are selected pathogenic defects. The high frequency of A7 T204fs mutations in MSI-positive cancers (22% overall) points to CTCF as a bona fide MSI target gene in endometrial cancers and accounts for the elevated mutation rate in MSI-positive tumors compared with tumors with intact DNA mismatch repair.
There is limited overlap between the mutations we observed and those described by TCGA that reported CTCF is a significantly mutated gene in both MSI-positive and microsatellite stable tumors: only five of the 24 different somatic mutations we observed were seen in the TCGA cohort (including p.T204fs*26 and p.T204fs*18 identified by Pindel). The large number of novel CTCF mutations speaks further to the importance of CTCF in endometrial tumorigenesis.
There is a growing number of genes involved in the regulation of chromatin structure for which somatic and constitutional defects have been associated with human malignancies and developmental abnormalities [Kandoth et al., 2013; van Bokhoven, 2011]. ARID1B is one such example. ARID1B is frequently mutated in endometrial cancers and constitutional mutations have been associated with syndromic mental retardation, including Coffin–Siris syndrome [Hoyer et al., 2012; Tsurusaki et al., 2012]. Recently, de novo frameshift CTCF mutations were associated with intellectual disability and it is hypothesized that CTCF deficiency affects expression of enhancer-regulated genes [Gregor et al., 2013]. The functional consequences of CTCF defects in endometrial cancer are yet to be determined and the analysis of missense mutations reported here and by the TCGA may be particularly revealing. Absence of CTCF binding in a chromatin boundary region upstream of the CDKN2A tumor suppressor is associated with p16 silencing [Witcher and Emerson, 2009]. Perturbed/reduced CTCF activity associated with missense mutations and/or haploinsufficiency could impact expression of a broad range of genes with known or novel roles in endometrial tumorigenesis.
Acknowledgments
We thank Matt O'Hern for assistance with mutation testing and Alexis Chassen for help with manuscript preparation. The study was approved by the Human Studies Committee at Washington University in St. Louis, MO and Human Subjects Research Committee at the Ohio State University in Columbus, OH. We are grateful to the patients who volunteered to participate in the study in order to make this work possible.
Disclosure statement: The authors declare no conflict of interest.

