

Journal list menu

 Issue
IssueJournal of Computational Chemistry: Volume 38, Issue 15
Special Issue:Simulating Biomolecules: Festschrift to commemorate the 60th birthday of Charles L. Brooks III.
i-ii, 1103-1282June 5, 2017
Export Citations
Download PDFs
Cover Image
Cover Image, Volume 38, Issue 15
- Page: i
- First Published: 24 April 2017

Cheri and Charlie in front of the Hallgrim's church in Reykjavik, Iceland, in May 2016. The original selfie was processed to this pencil drawing-like image by Sunyoung Kim. Charlie said “The research we, all of my former students, postdocs and collaborators and I, have accomplished is a partnership that builds on the strengths and experiences of each member. These partnerships, in my case, have mostly been long-lived and fruitful. My longest lived partnership and collaboration has been with Cheri, who has provided support, not just for me, but for most of those for whom I have had the privilege to work. She has been a stabilizing force of balance that has enabled me the freedom to pursue my scientific interests and is recognized here for her extraordinary contributions to the scientific advances represented in this volume”.
Cover Image, Volume 38, Issue 15
- Page: ii
- First Published: 24 April 2017

The discovery of allosteric modulators of G-protein coupled receptors (GPCRs) has led to significant interest in their therapeutic potential. On page 1209, (DOI: 10.1002/jcc.24728) Leon Sakkal, Kyle Rajkowski, and Roger Armen utilize recent structural information to model a series of positive allosteric modulators (PAMs) of adenosine and muscarinic acetylcholine receptors. This provides insight into how PAMs slow agonist radio ligand dissociation kinetics and exhibit probe-dependent activity. On the cover is a model of A3R PAM LUF6000 showing agonist specific interactions with inosine. This work has implications for the design of PAMs for Class A GPCRs, with potential use in myriad diseases, including stroke, myocardial infarction, Alzheimer's disease, and cancer.
Issue Information
Introduction
Simulating Biomolecules: Festschrift to commemorate the 60th birthday of Charles L. Brooks III
- Pages: 1111-1113
- First Published: 24 April 2017
Review
CHARMM-GUI 10 years for biomolecular modeling and simulation
- Pages: 1114-1124
- First Published: 14 November 2016

CHARMM-GUI, http://www.charmm-gui.org, is a web-based graphical user interface that can be used to prepare complex biomolecular simulation systems and input files (for CHARMM, NAMD, GROMACS, AMBER, GENESIS, LAMMPS, Desmond, OpenMM, and CHARMM/OpenMM) to facilitate the usage of common and advanced simulation techniques.
Full Papers
Molecular simulations to delineate functional conformational transitions in the HCV polymerase
- Pages: 1125-1137
- First Published: 12 November 2016
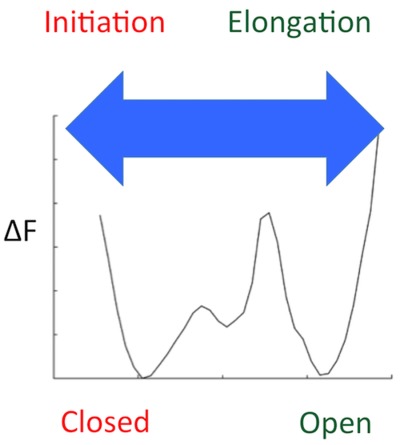
The Hepatitis C virus polymerase adopts distinct conformations in replicating the viral RNA genome. Molecular dynamics simulations reveal the physical nature of these states in previously unavailable detail, as well as the molecular interactions that allow the enzyme to transition between them. These studies provide invaluable information regarding the fundamental processes that govern RNA replication performed by the polymerase and contribute to improving our comprehension of molecular mechanisms by which therapeutics may target this enzyme.
Optimal allosteric stabilization sites using contact stabilization analysis
- Pages: 1138-1146
- First Published: 24 October 2016
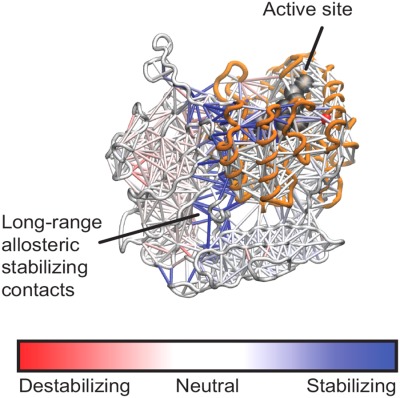
Proteins are prone to unfold and aggregate, especially when mutated or under stress. Stabilizing a proteins' functional state, either using engineered disulfide bonds or designed small-molecule stabilizers, is an important goal, but first we must determine an optimal stabilization site to impact activity. We use simulation to measure the stabilization capacity of every contact in the β-glucuronidase enzyme, and show that the stabilization capacity of a contact is only loosely related to its proximity from the active site.
The free energy of locking a ring: Changing a deoxyribonucleoside to a locked nucleic acid
- Pages: 1147-1157
- First Published: 19 January 2017
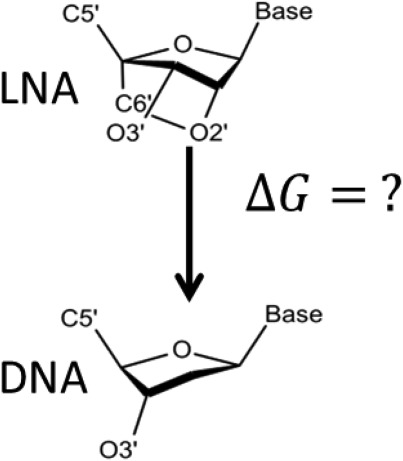
Locked nucleic acids (LNA) are widely used in gene therapeutic field, for its well-known high stabilization on DNA helical structures. While it is the interesting to calculate what the difference of its free energy contributions are from DNA, the technical difficulty of transforming a bridged-ring remains. This study describes the protocols of calculating free energy difference between LNA and DNA using MD simulations and Bennett Acceptance Ratio.
Insulin mimetic peptide S371 folds into a helical structure
- Pages: 1158-1166
- First Published: 12 February 2017
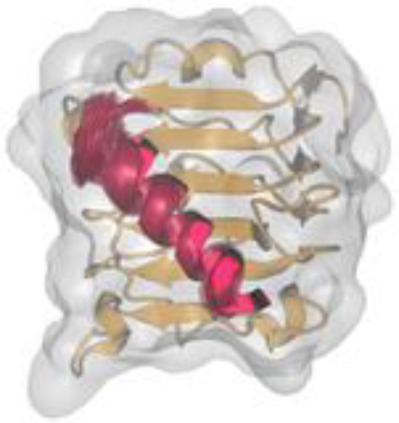
Insulin mimetic peptides have been recently designed as potent receptor agonists and antagonists. In this article are present structural models of a site 1 mimetic peptide (red transparent cartoons) that is known to compete with a critical hormone-binding helical peptide of the receptor (dark red cartoon) for binding at site 1 (brown cartoon and transparent surface).
Conformational changes of ubiquitin under high pressure conditions: A pressure simulated tempering molecular dynamics study
- Pages: 1167-1173
- First Published: 08 April 2017
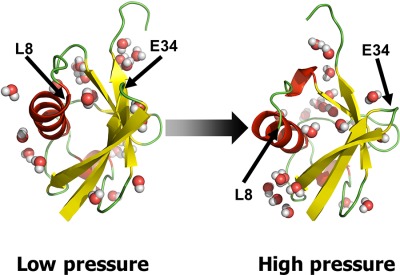
Our pressure simulated tempering molecular dynamics simulations for the system of ubiquitin show that the large fluctuations of the L8-E34 distance are induced by pressure. Ubiquitin at high pressure has a larger space between the upper side of the helix and the loop region opposite to the helix than at low pressure. There are more water molecules at high pressure in the region between the helix and the loop than at low pressure.
Extreme biophysics: Enzymes under pressure
- Pages: 1174-1182
- First Published: 19 January 2017
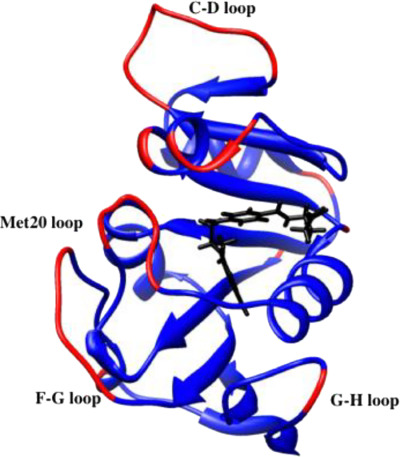
Determining adaptations of enzymes for extreme pressure and temperature is important for understanding structure-function relationships in enzymes and may help in defining the “limits of life.” Atomistic simulations of dihydrofolate reductase from a mesophile and a “piezophile” identify collective motions as responsible for the flexibility necessary for “corresponding states” enzyme activity. In addition, the adaptions for low temperature and high pressure environment of deep-sea microbes are identified as greater flexibility and lower density.
Effects from metal ion in tumor endothelial marker 8 and anthrax protective antigen: BioLayer Interferometry experiment and molecular dynamics simulation study
- Pages: 1183-1190
- First Published: 24 April 2017
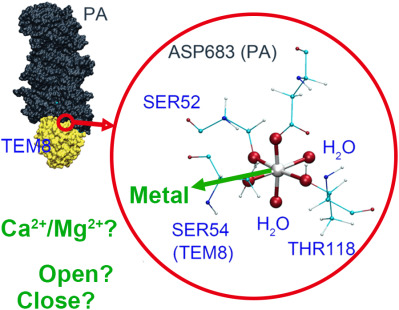
Tumor endothelial marker 8 (TEM8) is reported to bind to a potential anticancer target, protective antigen (PA), through an integral metal ion. Consistent with BioLayer Interferometry experiments, computer simulations indicate the metal ion contributes significantly to the binding affinity. TEM8-PA binding is more favorable in the presence of a smaller ion like Mg2+, rather than a larger ion like Ca2+. Moreover, the conformation change, which links to the change in activity of integrins, was not found in TEM8. In the presence of Mg2+, TEM8 remains in a conformation analogous to an integrin open (high-affinity) conformation.
Phage-like packing structures with mean field sequence dependence
- Pages: 1191-1197
- First Published: 27 March 2017
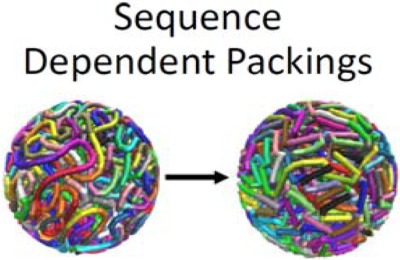
DNA sequence determines the response to packing stress in some phages. Left is a purely elastic model and right is a mean field sequence model with structural defects determined by the nonlinear elastic response to sequence thermodynamics.
Computing conformational free energy differences in explicit solvent: An efficient thermodynamic cycle using an auxiliary potential and a free energy functional constructed from the end points
- Pages: 1198-1208
- First Published: 23 December 2016
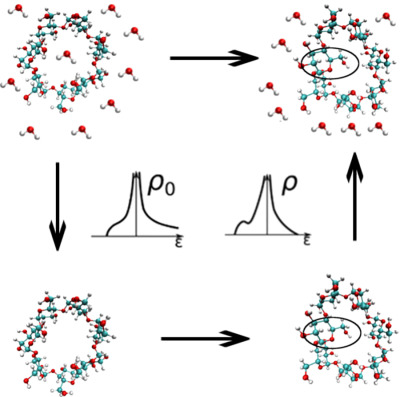
The free energy differences associated with conformational changes are difficult to compute in explicit solvent. Instead, these free energy differences can be computed on an auxiliary free energy surface and the desired free energy difference obtained by adding the free energies of transferring the end states from the auxiliary surface to the target surface. Here, we show that computing these transfer free energies with the energy representation method substantially reduces the cost of these calculations.
Prediction of consensus binding mode geometries for related chemical series of positive allosteric modulators of adenosine and muscarinic acetylcholine receptors
- Pages: 1209-1228
- First Published: 28 January 2017
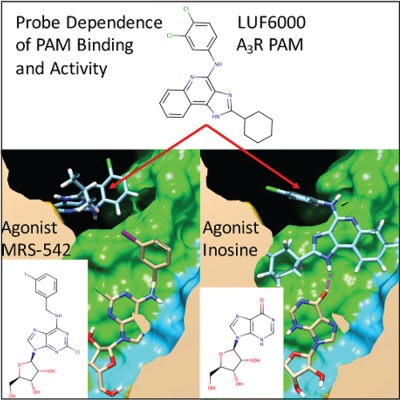
A3R partial-agonist inosine, in the presence of positive allosteric modulator LUF6000, exhibits three-fold increased activity. Docking studies predict LUF6000 binding in a “capping” interaction through a hydrogen bond with the carbonyl group of inosine. The interaction between the molecular structures of LUF6000 and inosine may explain the increase in activity. This interaction is inosine-specific, and is not seen with radioligand MRS-542, which lacks a carbonyl group.
Are induced fit protein conformational changes caused by ligand-binding predictable? A molecular dynamics investigation
- Pages: 1229-1237
- First Published: 16 April 2017
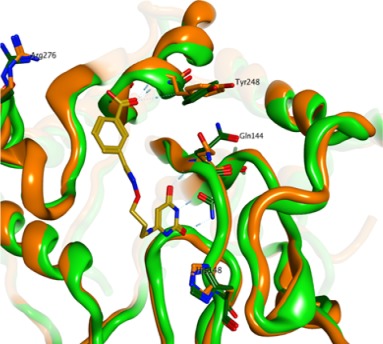
Conventional MD was run on 39 apo structures (no ligand), and the resulting trajectories were compared to a set of 147 holo X-ray structures (ligand-bound). The simulation results did not perform better than using the normalized crystallographic structural factors as predictors of active-site rigid residues (87% predictive power) and mobile residues (47% predictive power). These results suggest potential issues in the use of unligated simulation frames directly for drug design applications such as ligand docking.
Estimation of relative free energies of binding using pre-computed ensembles based on the single-step free energy perturbation and the site-identification by Ligand competitive saturation approaches
- Pages: 1238-1251
- First Published: 26 October 2016
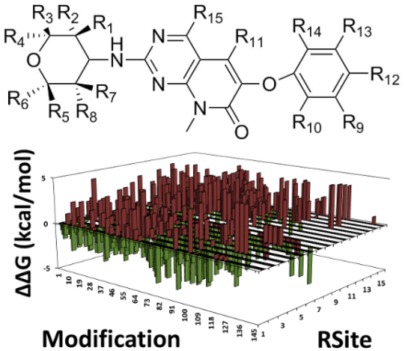
Accurate and rapid prediction of ligand potencies by the pre-computed ensembles based methods, Single Step Free Energy Perturbation (SSFEP) and Site Identification by Ligand Competitive Saturation (SILCS), is evaluated for Ack1 and p38 Map Kinase targets. Both SILCS and SSFEP are competitive with or better than the FEP results for the studied systems while being 1000+ fold times faster. Potential application of these methods in the context of screening large numbers of transformations is also illustrated.
On the importance of composite protein multiple ligand interactions in protein pockets
- Pages: 1252-1259
- First Published: 16 November 2016
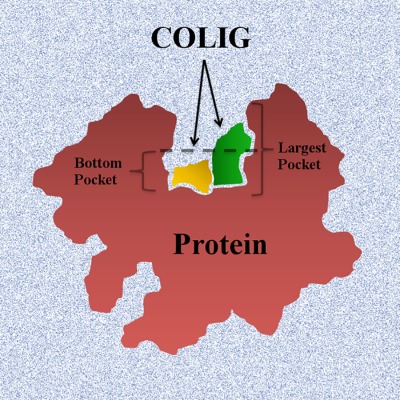
The canonical view of protein–ligand interaction is focused on one ligand per pocket at a time. Here, we present the concept of a COmposite Protein LIGands (COLIG), as multiple interacting ligands in the same pocket. Our analysis shows that majority of proteins can bind COLIGs and most ligands can form COLIGs. Finally, we find that ligands tend to bind in the deepest locations of the largest pocket of a protein.
Conformational dynamics of cathepsin D and binding to a small-molecule BACE1 inhibitor
- Pages: 1260-1269
- First Published: 02 April 2017
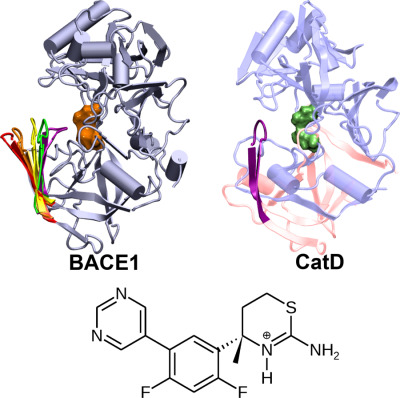
BACE1 is a major therapeutic target for Alzheimer's disease. Developing selective inhibitors has presented significant challenges. Here, we use continuous constant pH molecular dynamics simulations to investigate the conformational dynamics of cathepsin D (CatD) and its binding to a BACE1 inhibitor as a function of pH. Surprisingly, in the enzyme active pH range, the inhibitor forms a stronger salt bridge with the catalytic dyad in CatD than in BACE1, which might explain the off-target inhibition of CatD. Our work highlights the importance of incorporating pH effects in structure-based drug design.
A rapid solvent accessible surface area estimator for coarse grained molecular simulations
- Pages: 1270-1274
- First Published: 16 April 2017
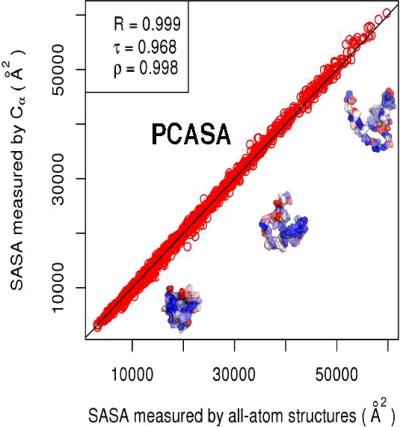
A fast and straightforward method is developed that predicts residue-wise solvent accessible surface areas (SASAs) from
 coordinates of protein structures. The method, Protein-C
coordinates of protein structures. The method, Protein-C
 Solvent Accessibilities or PCASA, should find utility as a tool for energetic and structural analysis of coarse-grained protein models. To ensure that the method was not biased toward native-like (folded) structures, PCASA is trained on a large dataset containing both folded and unfolded protein conformations. The resulting model is found to accurately recapitulate all-atom reference SASAs.
Solvent Accessibilities or PCASA, should find utility as a tool for energetic and structural analysis of coarse-grained protein models. To ensure that the method was not biased toward native-like (folded) structures, PCASA is trained on a large dataset containing both folded and unfolded protein conformations. The resulting model is found to accurately recapitulate all-atom reference SASAs.
PB-AM: An open-source, fully analytical linear poisson-boltzmann solver
- Pages: 1275-1282
- First Published: 02 November 2016

Visualization of electrostatic potential isosurface of charge-driven interaction between barnase (bottom left) and barstar (upper right) molecules, generated from the Poisson-Boltzmann Analytical Method (PB-AM) software. The PB-AM software is a user-friendly, fully analytical solver for the Poisson-Boltzmann equation.
Sign up for email alerts
Tools
