
Molecular Genetics & Genomic Medicine

Journal list menu

 Issue
IssueMolecular Genetics & Genomic Medicine: Volume 13, Issue 2
February 2025
Export Citations
Download PDFs
ISSUE INFORMATION
ORIGINAL ARTICLE
Unveiling the Genetic and Phenotypic Landscape of a Chinese Cohort With Retinitis Pigmentosa
- First Published: 23 February 2025
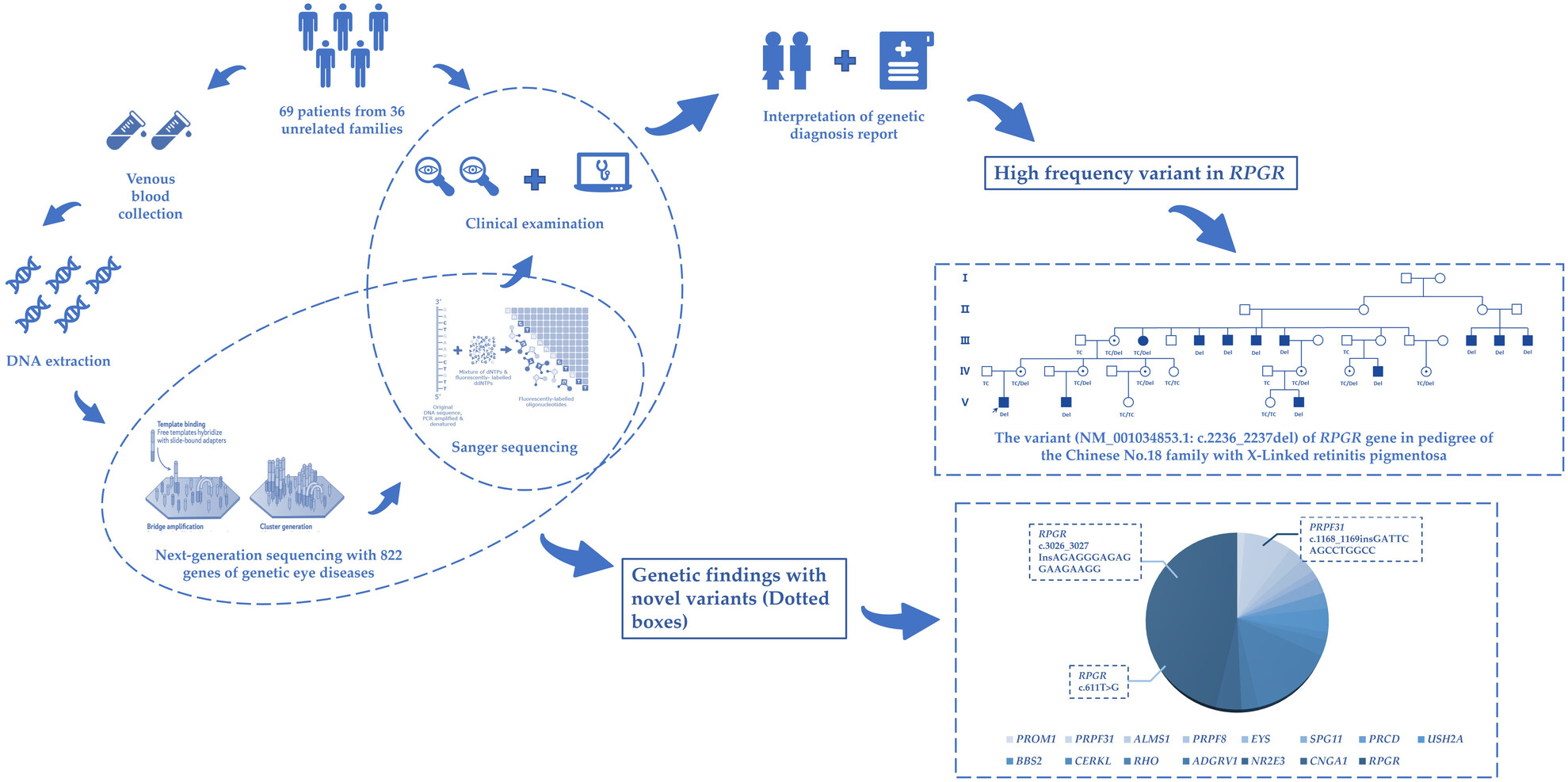
Our study conducted a genetic analysis of a cohort of 69 Chinese RP patients from 36 families. Genetic and phenotypic landscapes of this cohort were performed. Furthermore, three novel variants were identified in this study, and the high frequency variant (NM_001034853.1: c.2236_2237del) of RPGR detected from a big pedigree was discussed in detail, which may offer references for genetic diagnosis of retinitis pigmentosa.
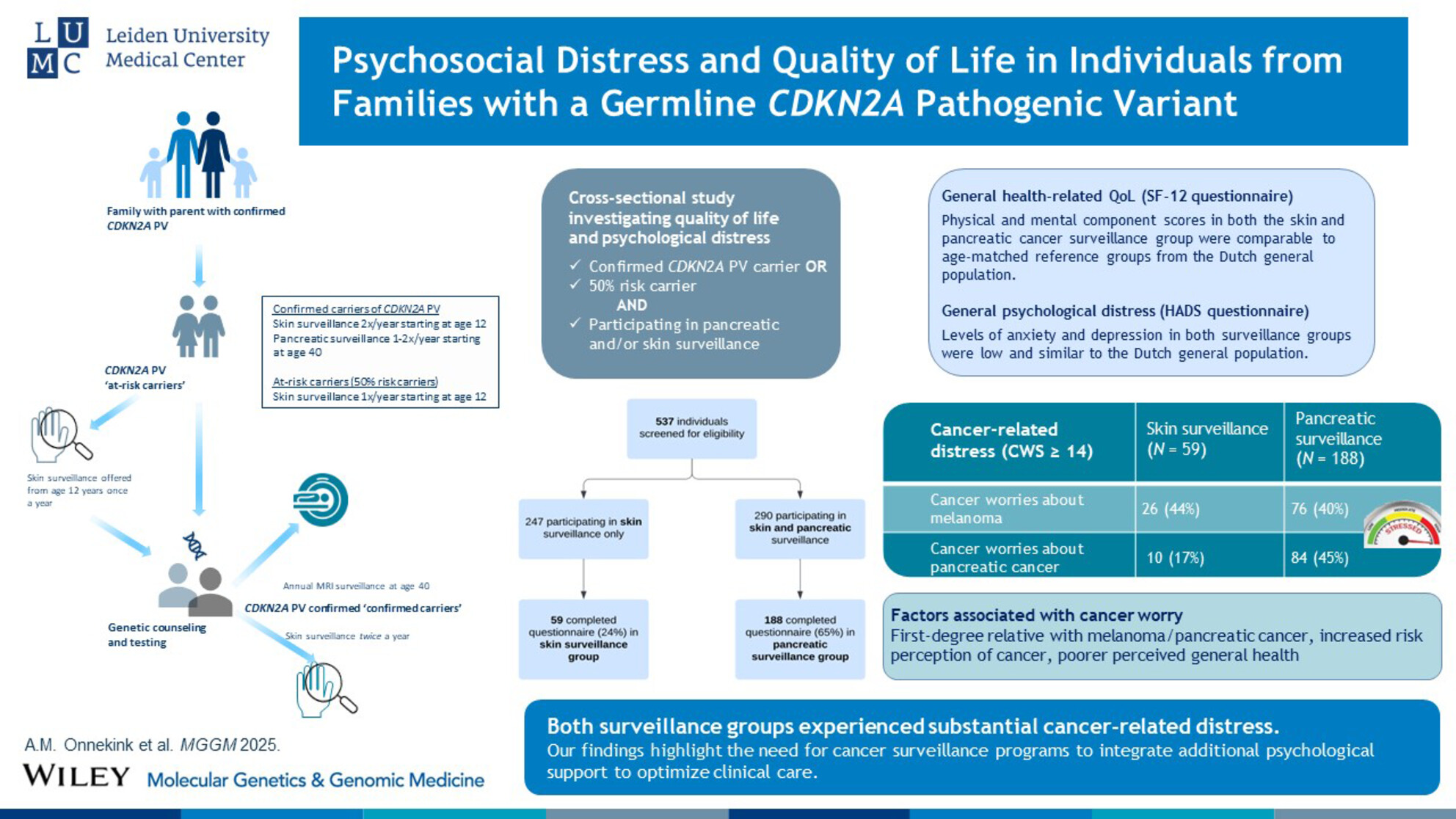
Psychological Distress and Quality of Life in Families With a Germline CDKN2A Pathogenic Variant
- First Published: 12 February 2025
Effects of a Novel COL4A3 Homozygous/Heterozygous Splicing Mutation on the Mild Phenotype in a Family With Autosomal Recessive Alport Syndrome and a Literature Review
- First Published: 09 February 2025
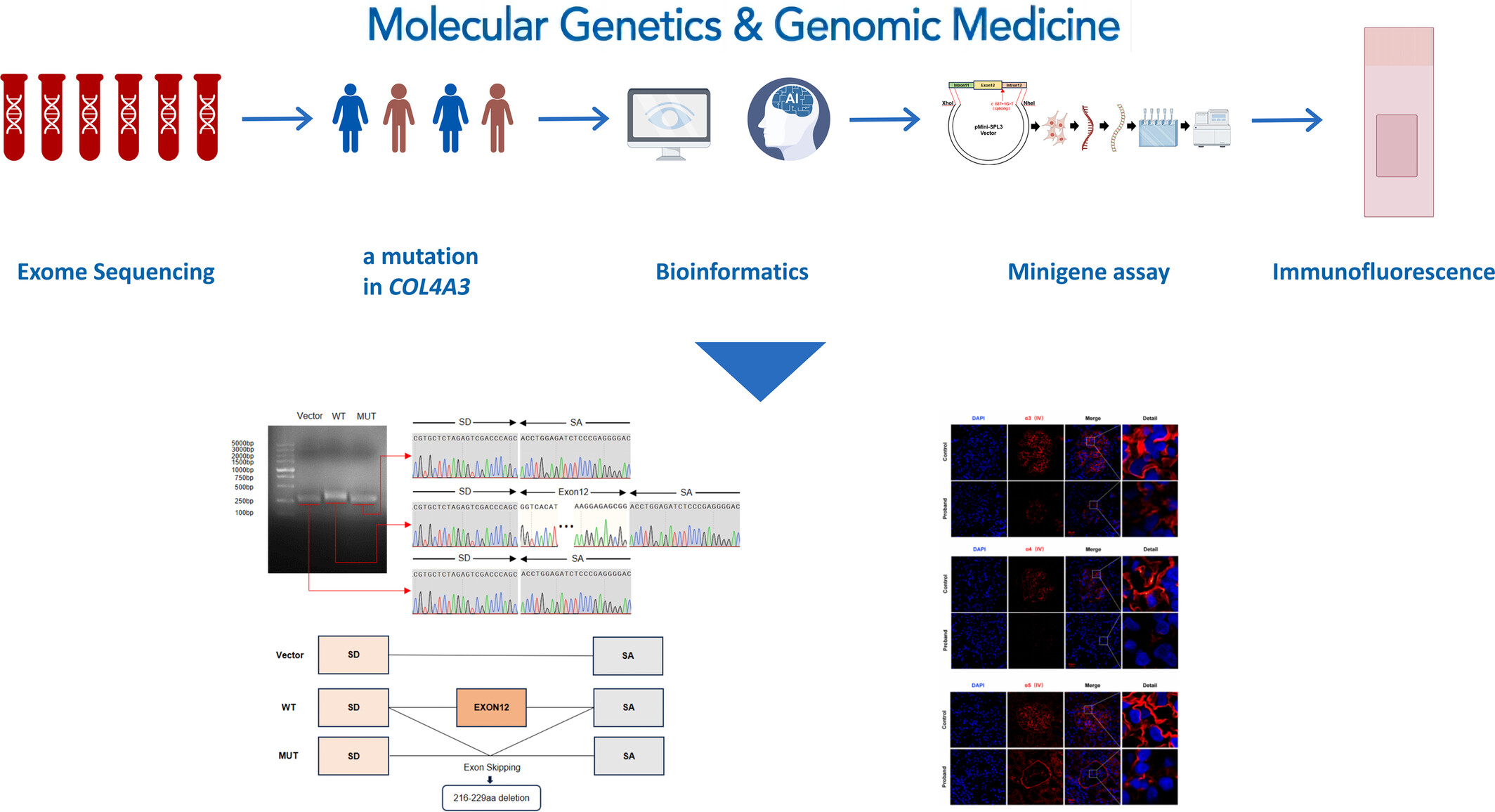
We present a novel finding of a previously unreported c.687 + 1G > T mutation in COL4A3, disrupting transcription and translation, impairing α3, α4, α5 (IV) chain formation. This alters the glomerular basement membrane integrity, causing hereditary Alport syndrome. This discovery enriches the genetic map of Alport syndrome, aiding clinical genetic guidance and enhancing pregnancy testing efficacy.
CLINICAL REPORT
Early Severe Cortical Involvement and Novel FUCA1 Mutations in a Pediatric Fucosidosis Case
- First Published: 25 January 2025
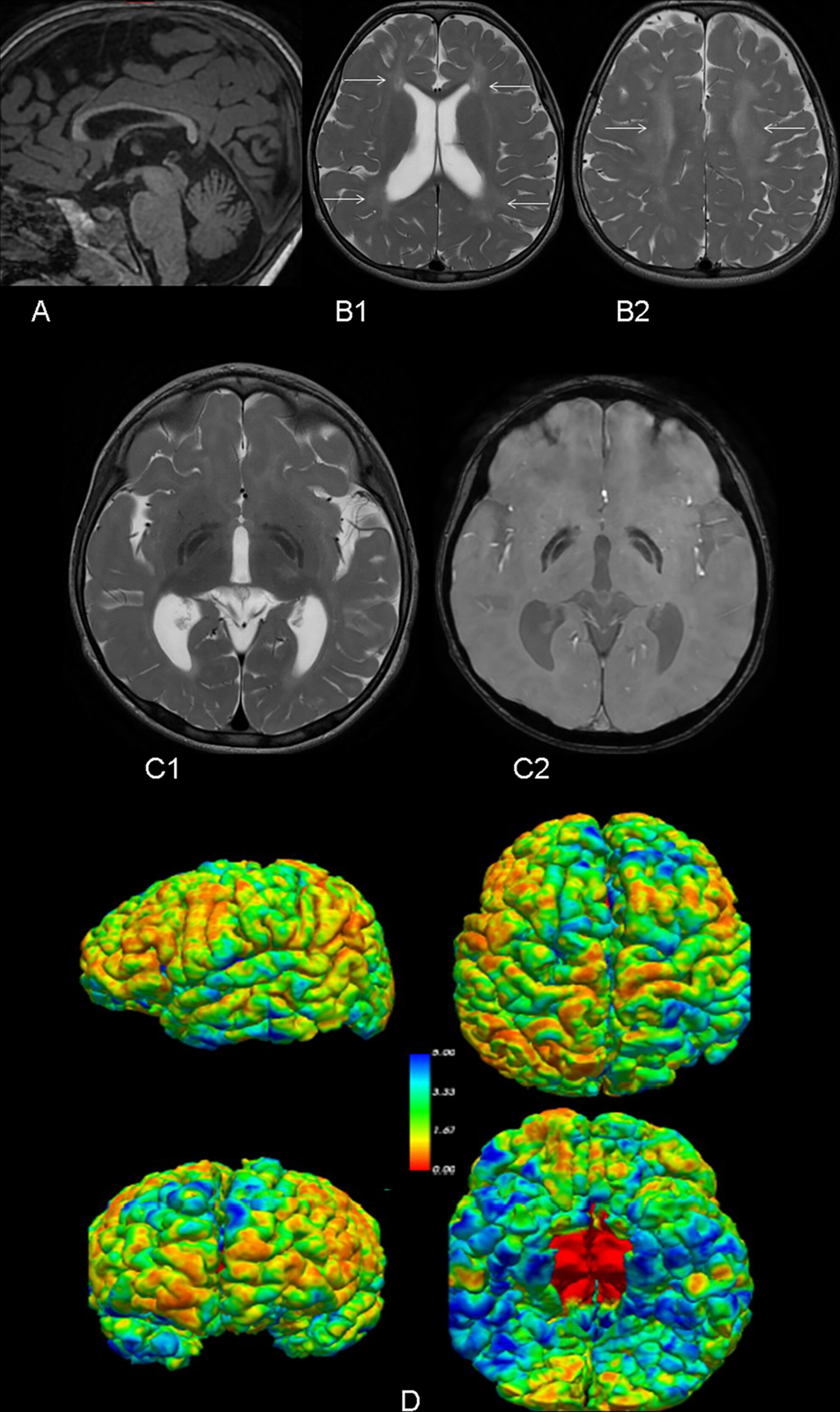
We report a 4-year-old boy with fucosidosis with novel pathogenic FUCA1 variants and neuroanatomical correlates. Results observed contribute to a better understanding of the genetics and pathophysiology of this rare lysosomal storage disorder.
A Novel Variant in TUBB4B Causes Progressive Cone-Rod Dystrophy and Early Onset Sensorineural Hearing Loss
- First Published: 29 January 2025
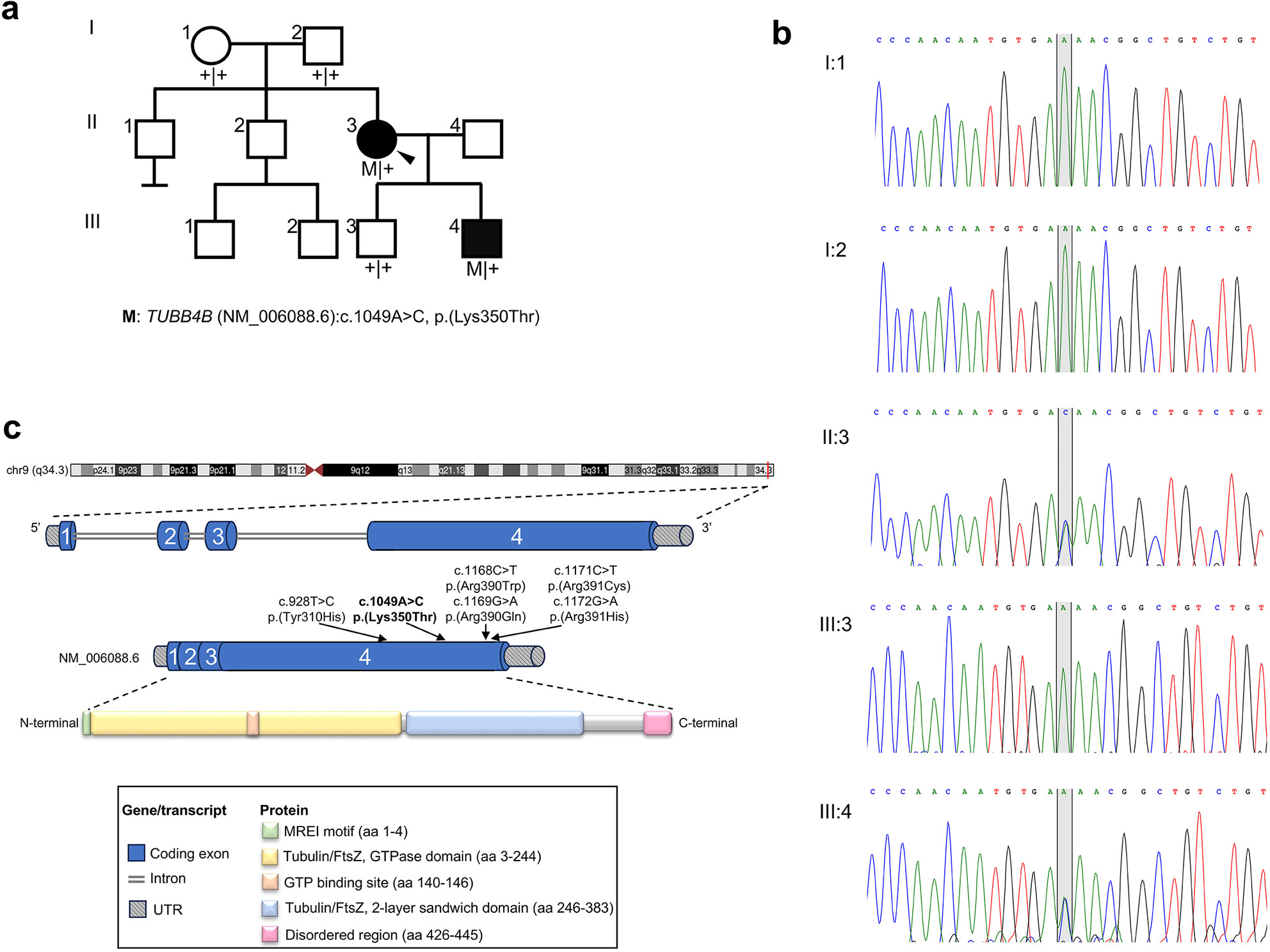
Whole-exome sequencing of affected members in a family segregating dominant syndromic retinal disease with early onset deafness revealed a novel, ultra-rare, disease-causing variant in TUBB4B (NM_006088:c.1049A>C). The variant substitutes a highly conserved lysine with threonine at amino acid position 350. Protein structure modeling studies suggest that Lys350 is an important residue for β-tubulin function, and detailed ophthalmological analyses demonstrate that the Lys350Thr TUBB4B variant causes progressive cone dystrophy with early onset sensorineural hearing loss.
Late-Onset Krabbe Disease: Case Report of Two Patients in a Chinese Family and Literature Review
- First Published: 29 January 2025
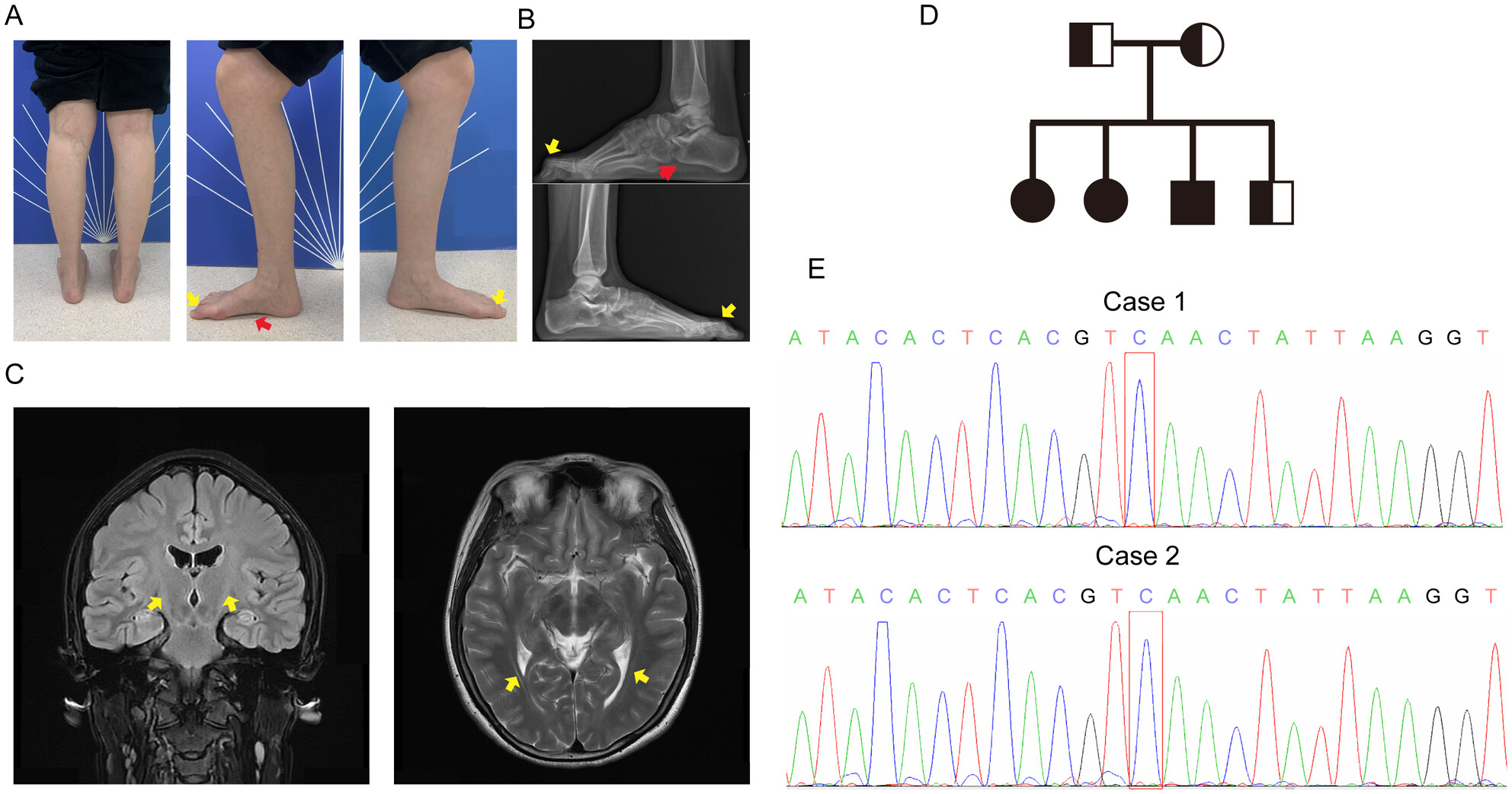
We report two cases of late-onset Krabbe disease (KD) from a Chinese family. This study broadens the consideration of KD in the diagnosis of patients presenting with muscle weakness and deformities in the lower limbs.
Rapid Whole Genome Sequencing Uncovers a Triple Diagnosis: X-Linked Chondrodysplasia Punctata, MECP2-Related Disorder, and Mosaic Jacobs Syndrome
- First Published: 05 February 2025

Neonate was found to have X-linked chondrodysplasia punctata, MECP2-related disorder, and mosaic Jacobs syndrome, highlighting the utility of advanced genetic testing in directing neonatal care and the complexity of managing multiple genetic diagnoses, while also adding to our understanding of the MECP2-related disorder phenotypes in boys.
ORIGINAL ARTICLE
Novel De Novo Intronic Variant of SYNGAP1 Associated With the Neurodevelopmental Disorders
- First Published: 29 January 2025
CLINICAL REPORT
Broadening the Phenotype Spectrum of MECP2 Variants in Men
- First Published: 30 January 2025
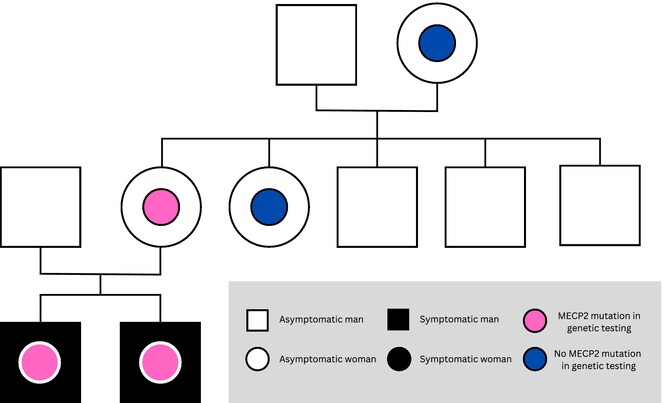
This case report presents a 23-year-old man and his younger brother who both carry maternally inherited hemizygous pathogenic missense variant c.419C>T, p.(Ala140Val) of MECP2. While the index patient has a severe syndromic intellectual disorder (ID), his outpatient brother has substantially milder ID predominantly limited to problems in linguistics skills.
ORIGINAL ARTICLE
The First Evidence for the Role of ACVR2A Gene Fetal Genotype in Preeclampsia Susceptibility
- First Published: 03 February 2025
The effect of fetal genotype on preeclampsia (PE) is usually missed in the literature. We showed the first evidence for the role of fetal ACVR2A polymorphisms in PE.
CLINICAL REPORT
A Novel Compound Heterozygous Variant in the ABHD12 Gene Cause PHARC Syndrome in a Chinese Family: The Proband Presenting New Genotype and Phenotype
- First Published: 05 February 2025
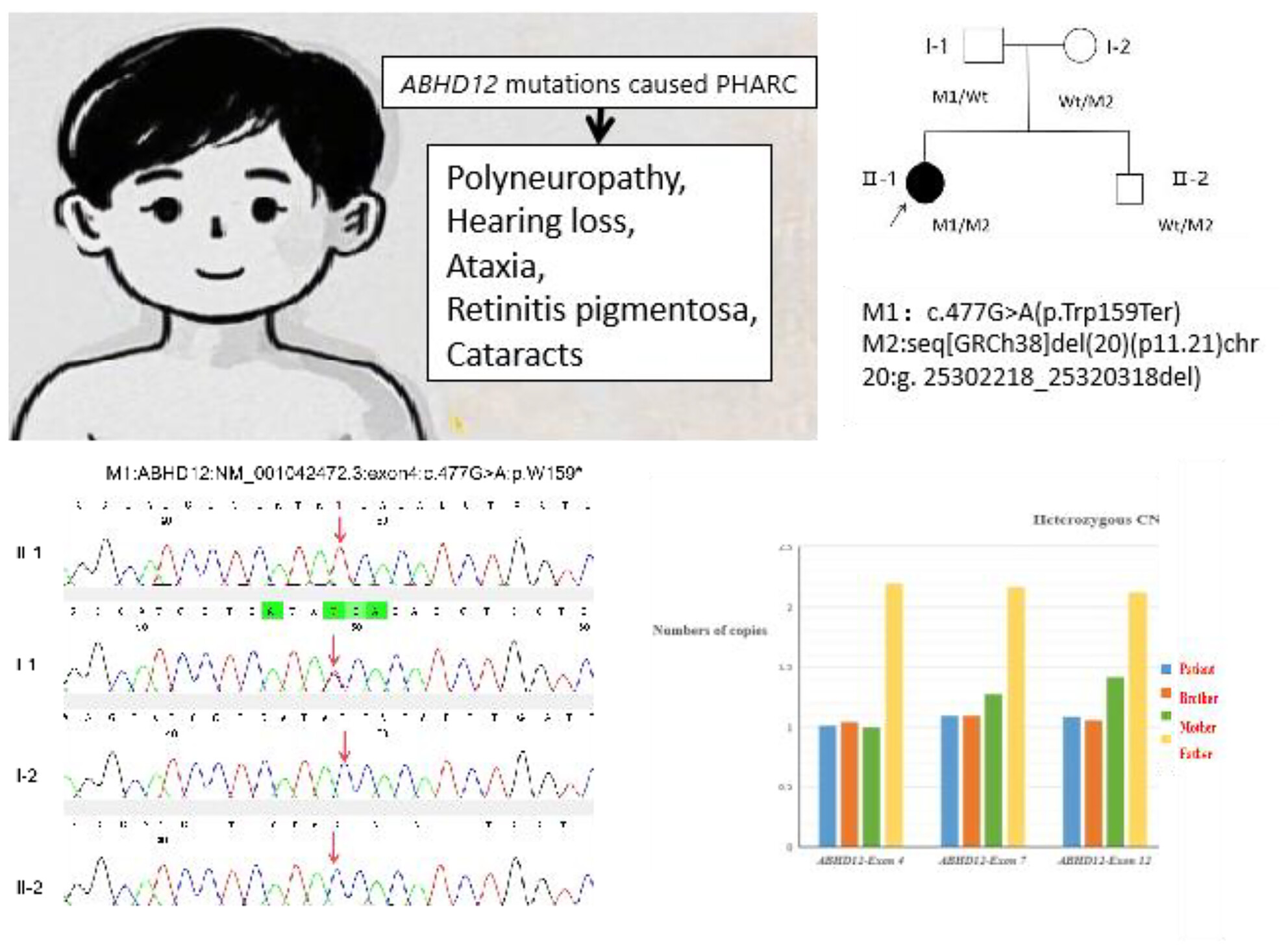
The principal symptoms of the proband comprised profound sensorineural hearing loss since childhood, severe visual impairment, congenital cataracts, cone-rod dystrophy, and ataxia. Whole Exome Sequencing (WES) revealed that the proband carried a compound heterozygous variant in the ABHD12 gene: M1, a known nonsense variation c.477G>A (p.Trp159Ter); and M2, a novel copy number variant with a deletion of approximately 18.10 Kbp in chromosome 20p11.21 (seq[GRCh38]del(20)(p11.21)chr20:g. 25302218_25320318del), covering exons 4-12 of the ABHD12 gene. The literature review indicated that there were 65 patients with PHARC from 30 different families. All clinical information of the described patients with PHARC syndrome and all known mutations associated with the disease to date were compiled.
CORRECTION
Correction to “Influence of the Sex of Translocation Carrier on Clinical Outcomes of Couples Undergoing Preimplantation Genetic Testing”
- First Published: 07 February 2025
CLINICAL REPORT
Novel De Novo RALA Missense Variants Expand the Genotype Spectrum of Hiatt-Neu-Cooper Neurodevelopmental Syndrome
- First Published: 07 February 2025
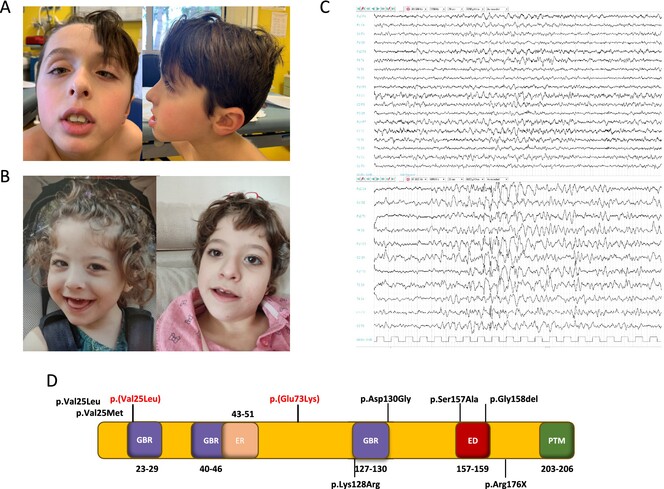
RALA is a small GTPase from the RAS superfamily involved in signal transduction and cytoskeletal dynamics. In this study, we identified two novel de novo missense variants in RALA (c.217G>A p.(Glu73Lys) and c.73G>C p.(Val25Leu)) in patients with developmental delay, epilepsy, and brain abnormalities. These rare variants affect conserved residues within the GTP/GDP-binding site, likely disrupting RALA's GTPase activity, which is crucial for neurodevelopment.
ORIGINAL ARTICLE
Reevaluation of the Impact of the Novel Likely Pathogenic Variant c.1286_1288delAGA in the ATP8A2 Gene: A 7-Year Follow-Up With Clinical, Genetic, and ACMG Insights in an Iranian Family
- First Published: 11 February 2025
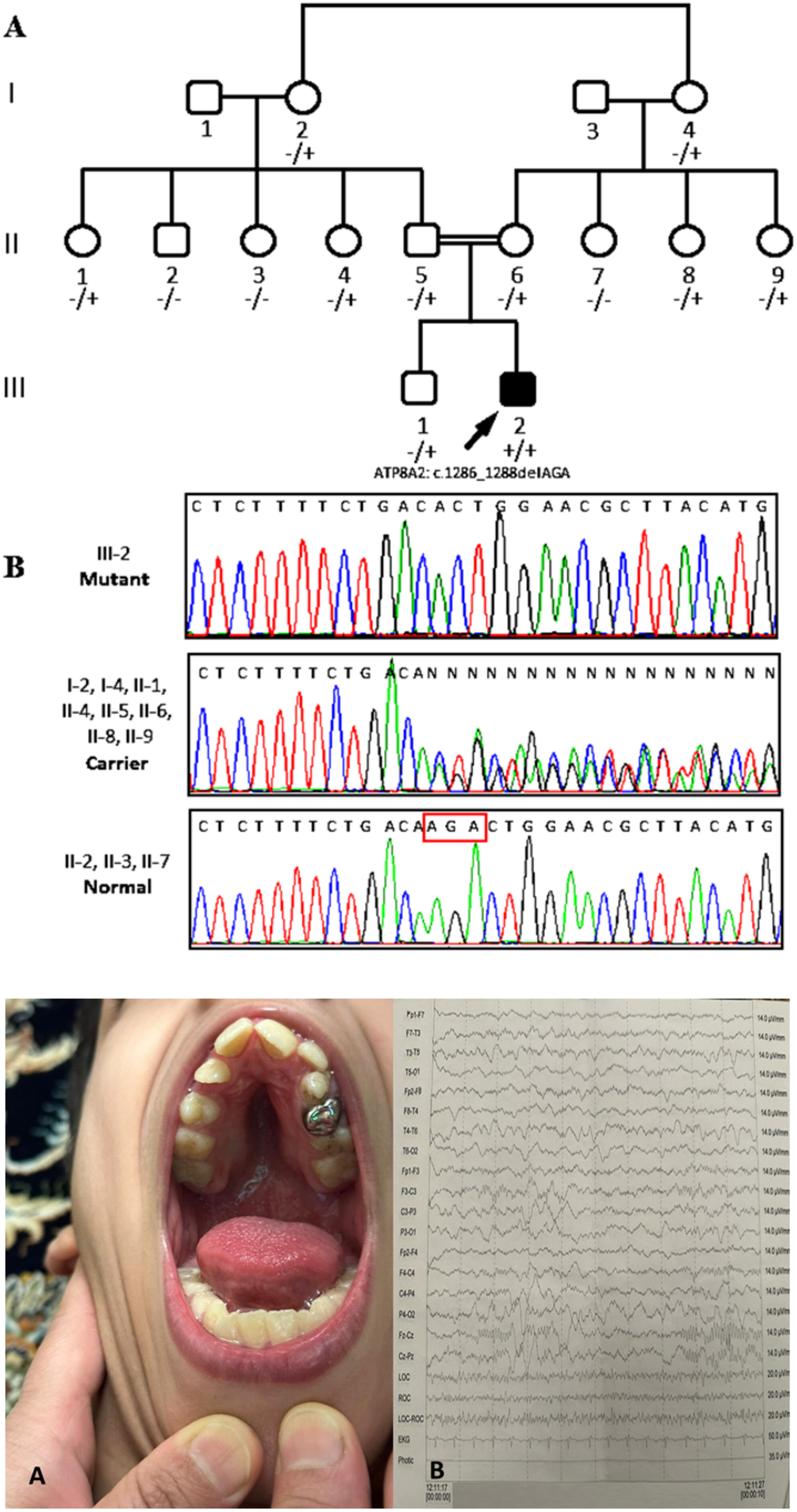
This study reports a 7-year follow-up of a male with CAMRQ4 syndrome caused by a homozygous in-frame deletion (c.1286_1288delAGA) in ATP8A2. Clinical and genetic analyses confirm its pathogenicity, expanding the mutational spectrum of ATP8A2-related CAMRQ and underscoring the importance of comprehensive genetic testing in rare neurological disorders.
CLINICAL REPORT
Spastic Paraplegia Type 78 Associated With ATP13A2 Gene Variants in Compound Heterozygosity
- First Published: 11 February 2025
In this work, a pathogenic variant screening was performed in a hereditary spastic paraplegia patient by whole-exome sequencing. Two potentially pathogenic ATP13A2 variants were selected and submitted to a segregation analysis with 16 family members, indicating that these variants are pathogenic for spastic paraplegia type 78 when occurring in compound heterozygosity.
ORIGINAL ARTICLE
The Genetics of 241 Fetuses With Talipes Equinovarus: A 8-Year Monocentric Retrospective Study
- First Published: 13 February 2025
Fetuses with talipes equinovarus (TE) is related to chromosome abnormalities, pathogenic or likely pathogenic copy number variants (CNVs) and monogenic diseases. Karyotyping, CMA, and WES could be offered. WES is recommended for both isolated and complex fetal TE.
REVIEW ARTICLE
The Functions and Implications of MicroRNAs in Premature Ovarian Insufficiency
- First Published: 17 February 2025
MiRNAs, distinct from protein-coding genes, play a critical role in the development of premature ovarian insufficiency (POI) by regulating various pathways such as epigenetic modifications, gene expression control, and participation in signaling cascades. This review explores the molecular mechanisms by which miRNAs influence POI pathogenesis and assesses their utility as biomarkers and therapeutic targets, potentially transforming early diagnosis, prognostic evaluations, and treatment approaches.
CLINICAL REPORT
Loss-of-Function CARS1 Variants in a Patient With Microcephaly, Developmental Delay, and a Brittle Hair Phenotype
- First Published: 18 February 2025

We report a patient with biallelic cysteinyl-tRNA synthetase (CARS1) variants that presents with hepatopathy, hypothyroidism, short stature, developmental delay, microcephaly muscular hypotonia, brittle hair, and ataxia. Functional studies revealed that the identified variants cause hypomorphic and loss-of-function effects, expanding the allelic and phenotypic heterogeneity of CARS1-associated disease.
ORIGINAL ARTICLE
Unraveling the Genetic Landscape of Hearing Loss: A Comprehensive Study of Azeri Families in Ardabil, Iran
- First Published: 18 February 2025
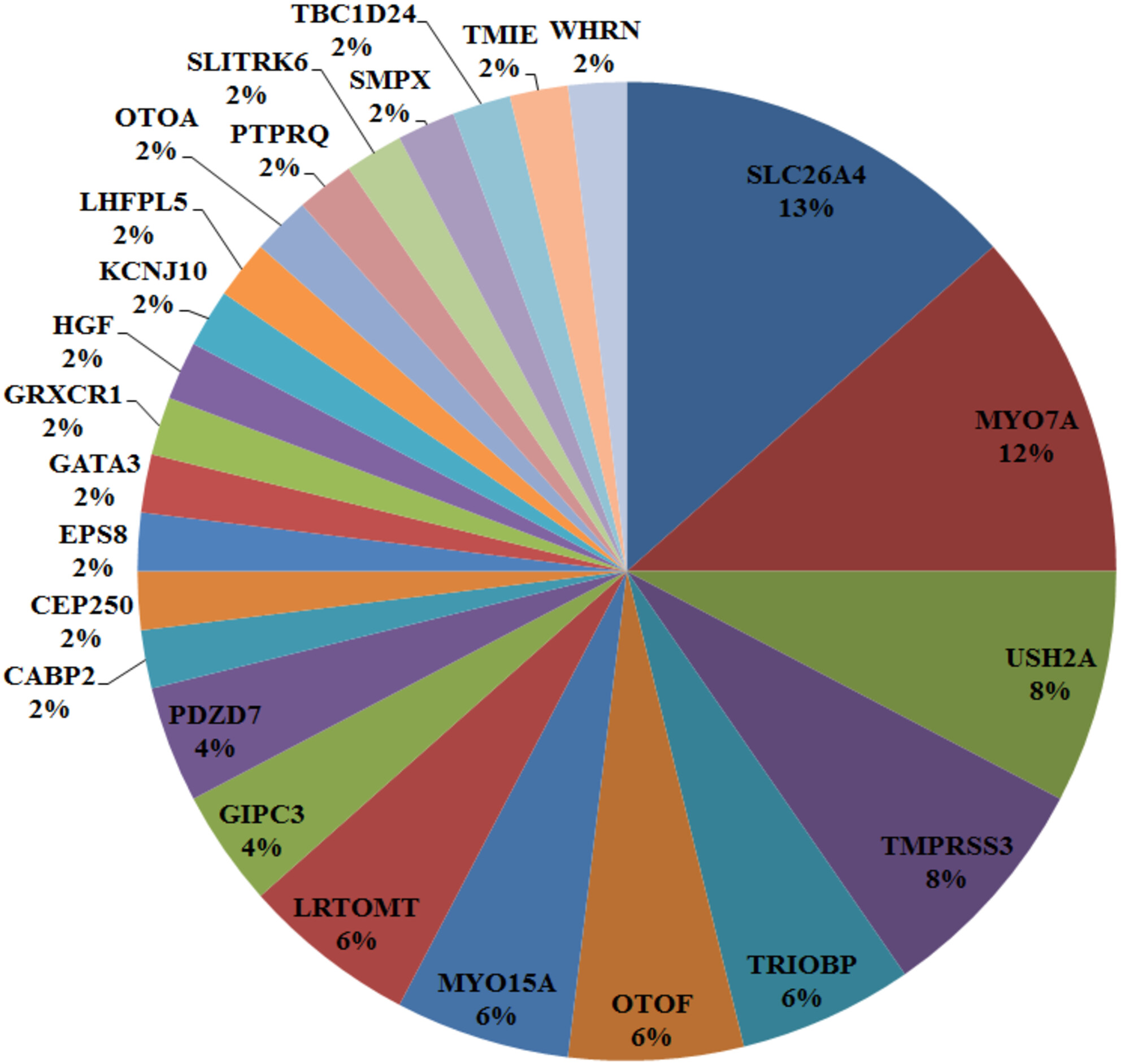
Our study highlights that SLC26A4 is the second most prevalent cause of hearing loss, following GJB2. This finding underscores the significance of understanding the genetic underpinnings of hearing loss for early diagnosis and the implementation of appropriate screening programs for different ethnic groups in Iran.
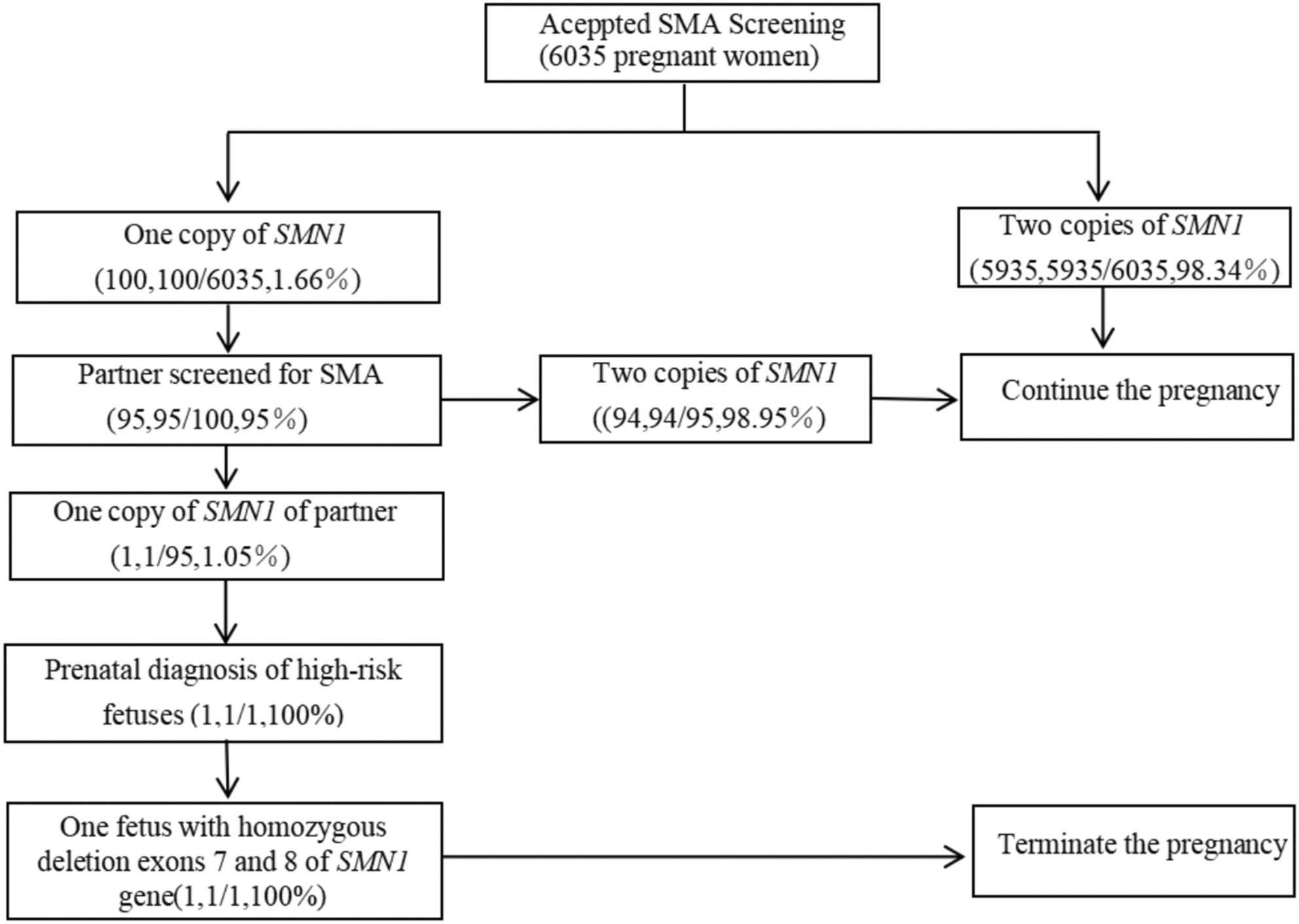
Carrier Screening and Prenatal Diagnosis for Spinal Muscular Atrophy in Ningde City, Fujian Province
- First Published: 19 February 2025
Trichothiodystrophy due to ERCC2 Variants: Uncommon Contributor to Progressive Hypomyelinating Leukodystrophy
- First Published: 20 February 2025

This study reports a novel case of trichothiodystrophy (TTD) linked to compound heterozygous ERCC2 variants, presenting with progressive cerebral hypomyelination. The findings highlight a rare association between ERCC2 mutations and hypomyelinating leukodystrophy, expanding the current understanding of TTD-related neurodevelopmental disorders.
Unmasking a Recessive Allele by a Rare Interstitial Deletion at 10q26.13q26.2: Prenatal Diagnosis of MMP21 -Related Disorder and Further Refine INSYN2A Involvement in the Postnatal Cognitive Phenotype
- First Published: 20 February 2025
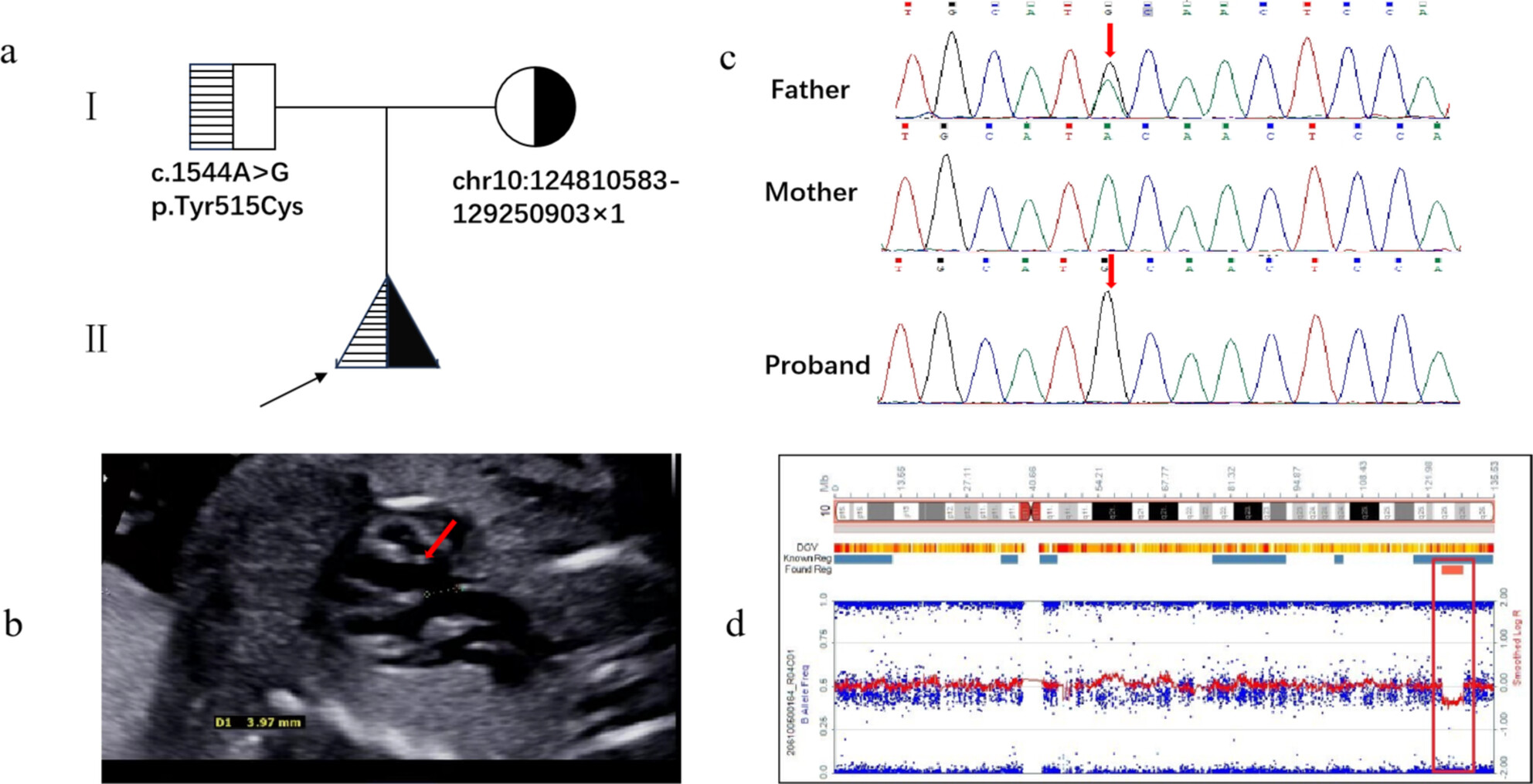
This study introduces a recessive MMP21 mutation unmasked by a rare 10q26.13q26.2 deletion via WES in a Chinese fetus and further refines INSYN2A as a potential candidate gene for cognitive phenotype in 10q26.1-q26.3 region, providing a reference for prenatal diagnosis and genetic counseling in patients with similar conditions.
Saliva Sample-Based Non-Invasive Carrier Screening for Spinal Muscular Atrophy, Hereditary Hearing Loss, and Thalassemia in 13,926 Women of Reproductive Age From South Zhejiang
- First Published: 23 February 2025
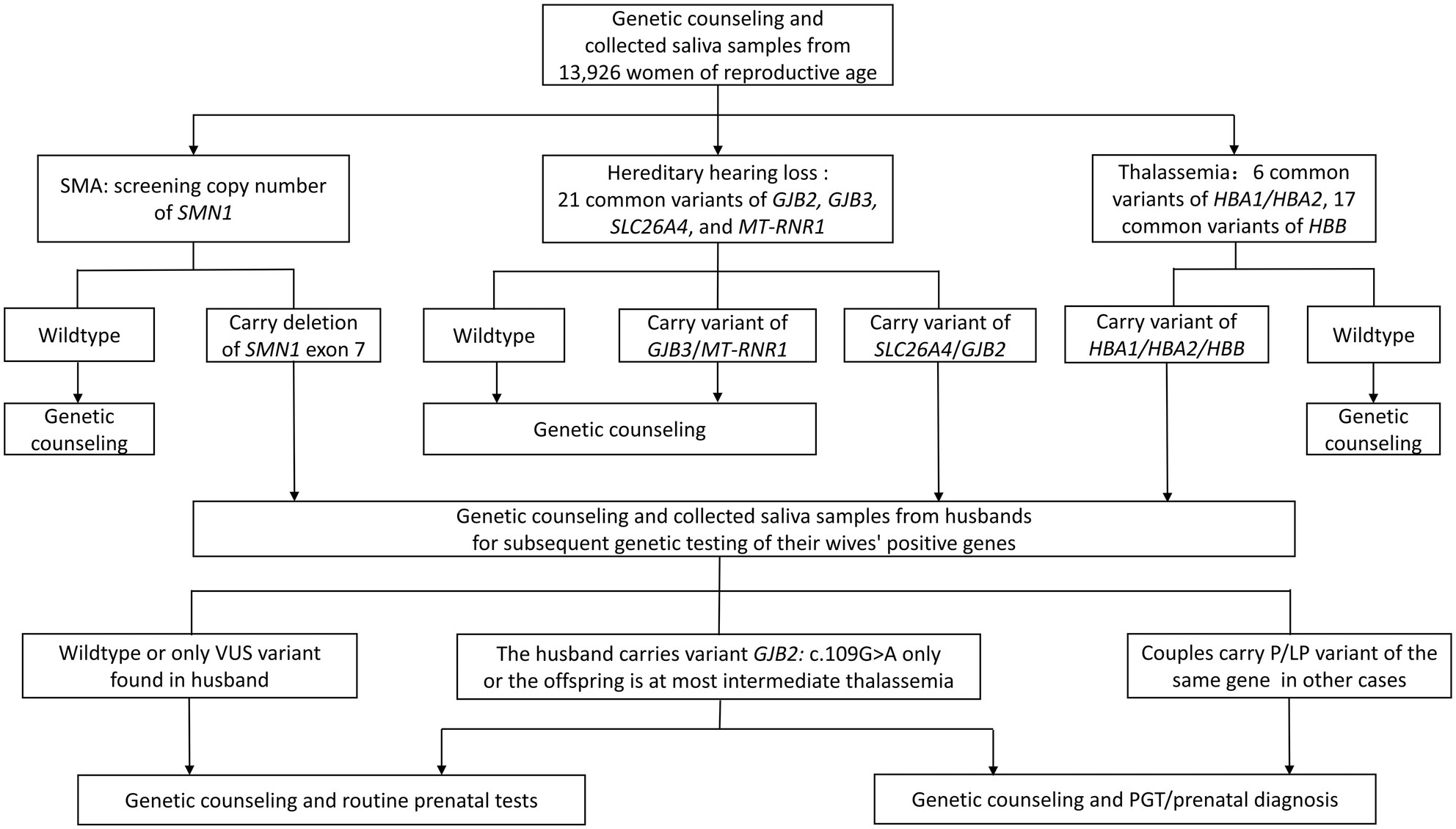
We established a rapid saliva-based non-invasive screening model for carriers of SMA, hearing loss, and thalassemia among women from South Zhejiang and provided prenatal diagnosis for high-risk families. The results not only clarified the local carrier frequency of these diseases but also confirmed the feasibility of the carrier screening model.
Identification and Validation of Biomarkers in Metabolic Dysfunction-Associated Steatohepatitis Using Machine Learning and Bioinformatics
- First Published: 24 February 2025
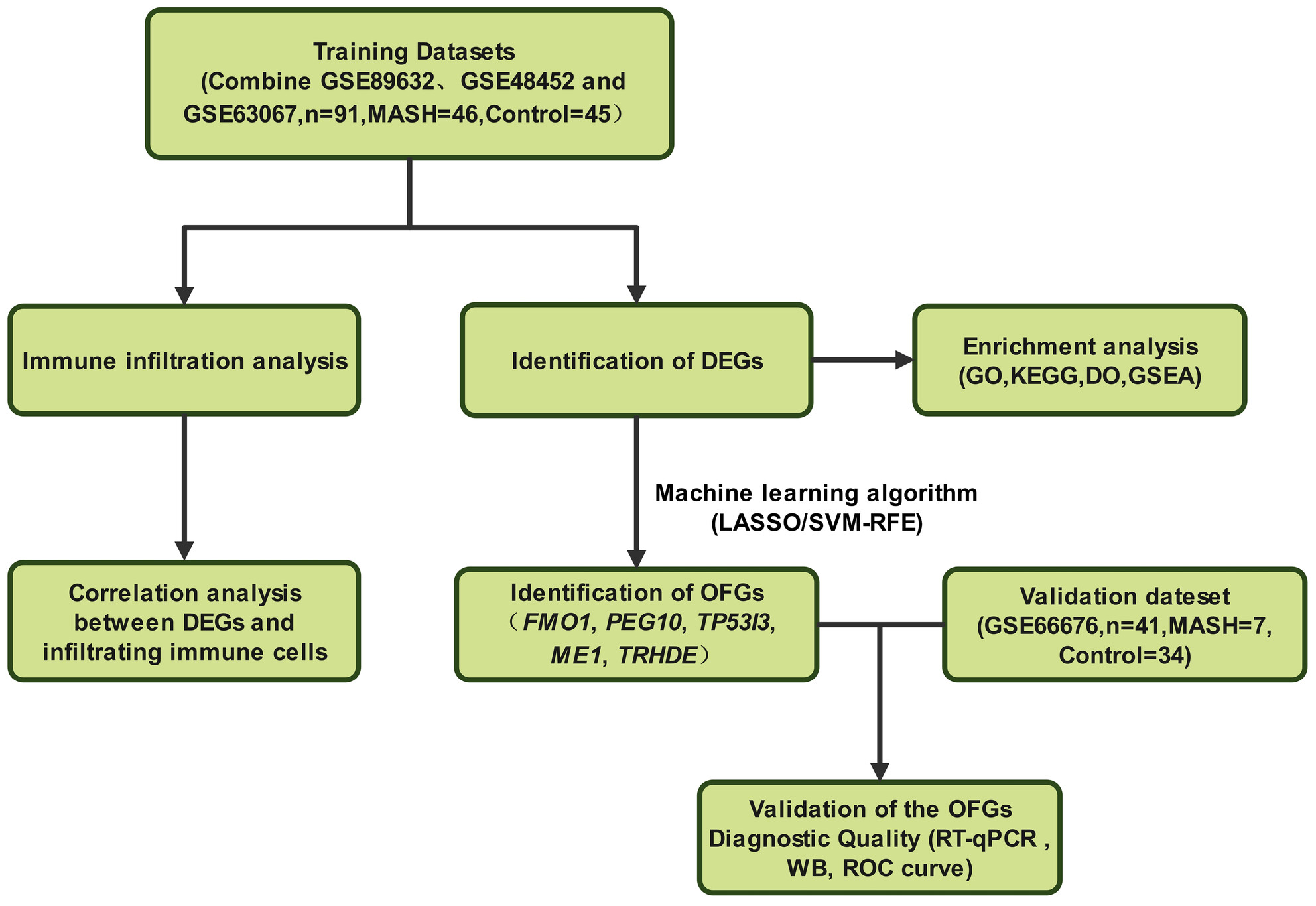
In this study, we used the Gene Expression Omnibus and applied the LASSO and SVM-RFE algorithms to identify differentially expressed genes (DEGs) in patients with metabolic dysfunction-associated steatohepatitis (MASH), aiming to identify diagnostic biomarkers or new therapeutic targets for MASH. The expression of these genes were validated in both in vitro and in vivo liver lipotoxicity models, confirming their alignment with the predictions. Five DEGs (FMO1, PEG10, TP53I3, ME1, and TRHDE) were initially identified by screening MASH and healthy control groups. Among these, ME1 exhibited an AUC exceeding 0.88 in testing and validation datasets, suggesting its potential as a predictive marker for MASH.




