

Journal list menu

 Issue
IssueInternational Journal of Quantum Chemistry: Volume 116, Issue 21
Special Issue:Theoretical and Computational Chemistry in Italy
i, 1493-1611November 5, 2016
Export Citations
Download PDFs
Cover Image
Cover Image, Volume 116, Issue 21
- Page: i
- First Published: 16 September 2016

On page 1575, Matteo Bonfanti and Rocco Martinazzo examine reactions at surfaces under a magnifying glass. Fruitful combination of theory, modelling, and simulations provides a powerful tool to understand the dynamics of atoms and molecules at the gas-solid interface. This is pictured on the cover with a simple illustration of two prototypical recombination processes, the Eley-Rideal (left) and the Langmuir-Hinshelwood (right) reactions. Image credit goes to Matteo Bonfanti. (DOI: 10.1002/qua.25192)
Issue Information
Editorial
Theoretical and computational chemistry in Italy
- Pages: 1499-1500
- First Published: 16 September 2016
Perspective
From oxide to proton conduction: A quantum-chemical perspective on the versatility of Sr2Fe1.5Mo0.5O6−δ-based materials
- Pages: 1501-1506
- First Published: 27 May 2016
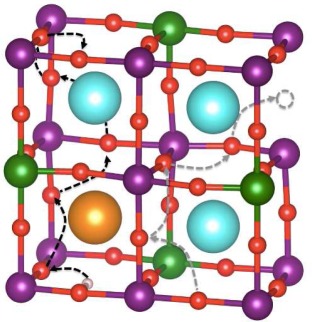
Quantum-chemical investigations can boost the development of advanced materials for energy conversion technologies. In the context of solid oxide fuel cells, the case of Sr2Fe1.5Mo0.5O6−δ-based electrodes exemplifies the successful application of DFT methods to the rational design of triple-conductor oxides, highlighting the key structure–property–function relationships that determine the oxide and proton bulk transport processes in perovskite oxides.
Computation modeling as a tool for the exploration of complex multistep reaction cycles in homogeneous catalysis. Some selected examples in the framework of the use of hydrogen as a fuel of the future
- Pages: 1507-1512
- First Published: 08 July 2016
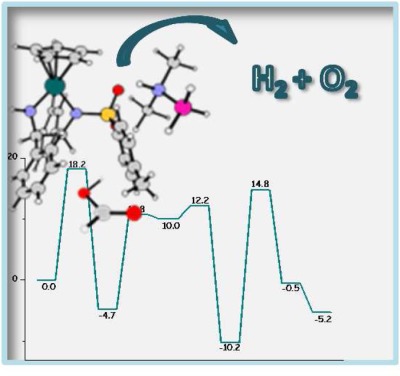
Computational modeling methods play a prominent role in the field of homogeneous catalysis given that reaction cycles tend to be multistep complicated processes, where the active catalytic species or intermediates are challenging or impractical to study via experimental approaches. The quantum mechanical description of intricate catalytic cycles is discussed here for three homogeneous catalytic systems all focused on the use of hydrogen as a potential zero-emission energy carrier for the future.
Accurate molecular structures of small- and medium-sized molecules
- Pages: 1513-1519
- First Published: 09 July 2016
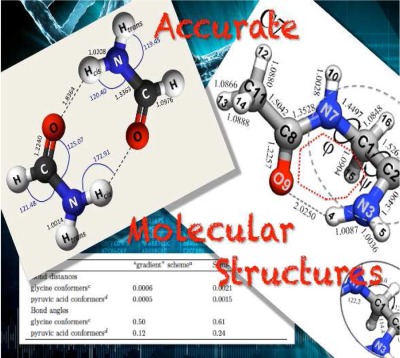
The development of computational strategies that permit accurate equilibrium structure determinations for systems ranging from small molecules to medium-sized biosystems is one of the main challenges in theoretical chemistry. Composite approaches have been developed in order to account for both electron correlation and basis-set effects at the best possible level. The additivity approximation is at the heart of these approaches: the various contributions are evaluated separately at the highest possible level and then combined together.
Reviews
Recent advances in solid-state NMR computational spectroscopy: The case of alumino-silicate glasses
- Pages: 1520-1531
- First Published: 26 March 2016
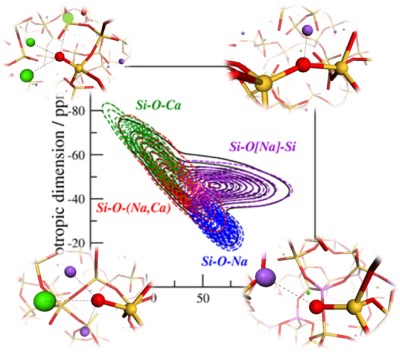
Molecular dynamics simulations coupled with DFT-GIPAW NMR calculations and spin-effective Hamiltonians provide a clear view of the local and medium range structure of multicomponent alumina silicate glasses.
Integrated QM/polarizable MM/continuum approaches to model chiroptical properties of strongly interacting solute–solvent systems
- Pages: 1532-1542
- First Published: 09 July 2016
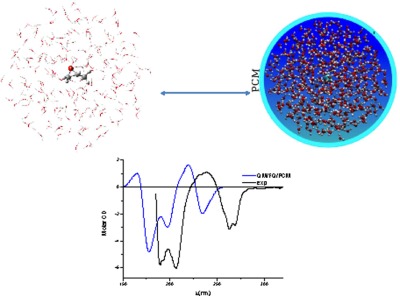
Solvent effects have great influence on chiroptical properties of molecular systems, and therefore cannot be ignored in theoretical models. Recently developed, integrated QM/polarizable MM/Continuum strategies based on fluctuating charges have demonstrated to be powerful approaches to quantitatively reproduce chiroptical properties and spectroscopies of aqueous solutions dominated by specific hydrogen-bonding effects.
Tutorial Reviews
Aiming at an accurate prediction of vibrational and electronic spectra for medium-to-large molecules: An overview
- Pages: 1543-1574
- First Published: 13 July 2016
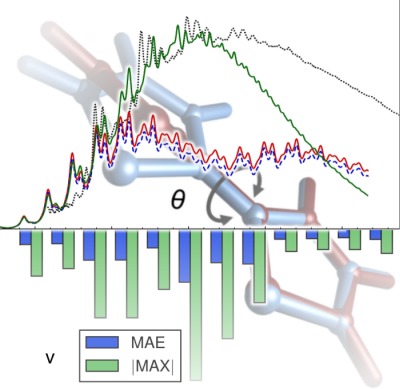
Cost-effective methodologies to simulate and interpret accurate vibrational and electronic spectra, directly comparable to experiment, are described from a theoretical and practical perspectives. Key aspects regarding their application on real cases are discussed through the design of a complete computational protocol starting from the definition of the best suited electronic structure calculation method, using thiophene and bithiophene as test cases. Possible shortcomings and strategies to overcome the limitations of those methods are also presented.
Classical and quantum dynamics at surfaces: Basic concepts from simple models
- Pages: 1575-1602
- First Published: 07 July 2016
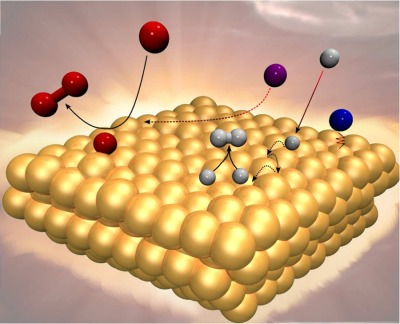
Simple classical and quantum analytical models provide valuable insight into elementary dynamical processes occurring at the gas-surface interface. In particular, information can be obtained from calculations on energy transfer processes that determine the establishment of an adsorbed phase on the surface, as well as the efficiency and the energy partitioning of most gas-surface reactions.
Software News & Updates
A new time-dependent density-functional method for molecular plasmonics: Formalism, implementation, and the Au144(SH)60 case study
- Pages: 1603-1611
- First Published: 16 June 2016
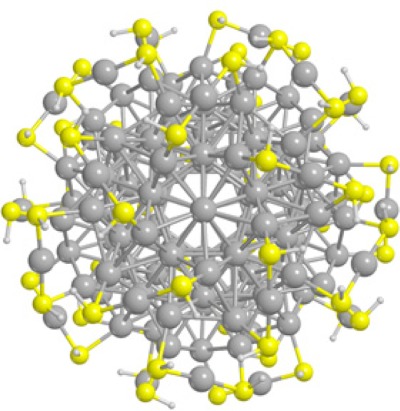
Efficient and accurate prediction of photoabsorption spectra for large systems is a fundamental goal of modern computational research. This is usually achieved within the time-dependent density-functional theory (TDDFT) approach but high associated computational costs limit its applicability to large systems. A recently developed TDDFT algorithm based on the complex dynamical polarizability to calculate the optical photoabsorption of large metal clusters is particularly suitable for applications in the field of plasmonics.
Sign up for email alerts
Tools
