

Journal list menu

 Issue2013v, 927-1047
Issue2013v, 927-1047
Export Citations
Download PDFs
Different Molecular Consequences of Frameshift Mutations in the ANTXR2 Gene
- Page: v
- First Published: 18 June 2013
Tetrahydrobiopterin, its Mode of Action on Phenylalanine Hydroxylase, and Importance of Genotypes for Pharmacological Therapy of Phenylketonuria
- Pages: 927-936
- First Published: 04 April 2013
Tetrahydrobiopterin (BH4; Kuvan®) induces a gain-of-function in phenylalanine hydroxylase (PAH) in a multifactorialresponse mechanism with a notable stabilization of protein structure. In about 20%-30% of phenylketonuria patients (all phenotypes of PAH deficiency), Phelevels may be controlled through PAHcofactor BH4 therapy. A substantial residual PAH activity is essential for the stabilization of enzyme activity and depends on specific PAH gene variations and on interallelic complementation, as shown from in vitro co-expression experiments.
Online Biomedical Resources for Malaria-Related Red Cell Disorders
- Pages: 937-944
- First Published: 08 April 2013
This paper reviews the content of open access biomedical databases including data on malaria-related red cell disorders launched over the last two decades, most of which focus on genetic diversity, and describes a new epidemiological resource developed by the Malaria Atlas Project. This resource currently includes data on hemoglobin S, hemoglobin C, the Duffy blood group, and G6PD deficiency and paves the way to similar work on the thalassemias.
Simple and Efficient Identification of Rare Recessive Pathologically Important Sequence Variants from Next Generation Exome Sequence Data
- Pages: 945-952
- First Published: 28 March 2013
Next generation sequencing (NGS) has quickly become the method of choice for seeking pathogenic mutations in rare uncharacterized monogenic diseases. However, the analysis of this data is challenging for small groups; consequently, we have developed a user friendly suite of programs that can interactively analyze, filter, and screen data from enrichment-capture NGS data.
A Rare Motor Neuron Deleterious Missense Mutation in the DPYSL3 (CRMP4) Gene is Associated with ALS
- Pages: 953-960
- First Published: 08 April 2013
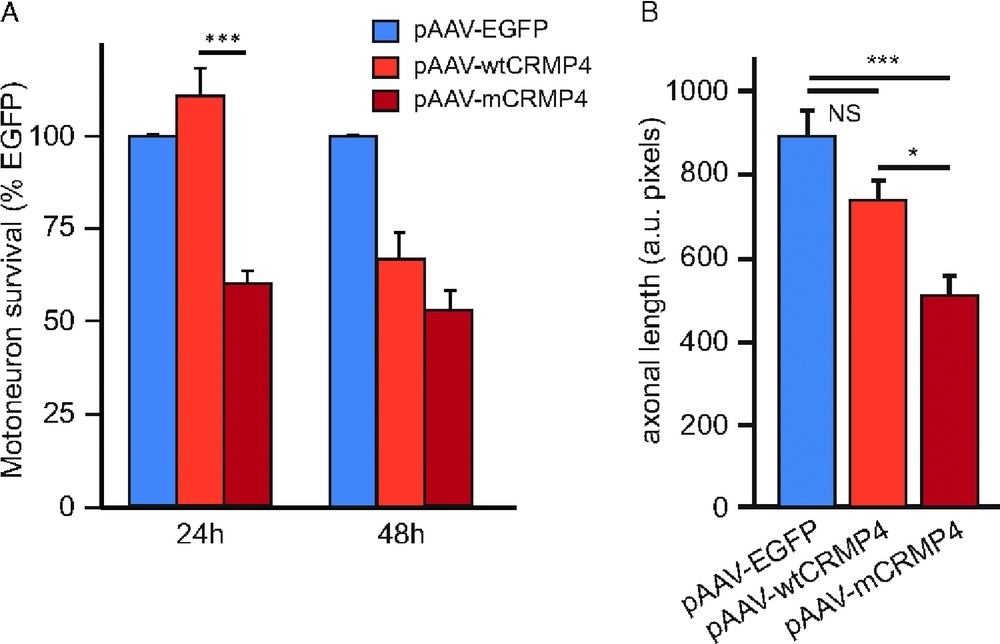
Twenty-four hours after electroporation, motor neuron survival was decreased to 58 ± 4% (P < 0.001) of expected when expressing mCRMP4 compared with wtCRMP4 or EGFP. Axonal growth, evaluated at 20 hr, was also reduced by 62 ± 10% (P < 0.001) in motor neurons expressing the mutated form of CRMP4 compared with EGFP expressing cells, whereas expression of the wildtype form of CRMP4 did not significantly affect axonal growth (87 ± 7%)
A Missense Mutation in the Sodium Channel β2 Subunit Reveals SCN2B as a New Candidate Gene for Brugada Syndrome
- Pages: 961-966
- First Published: 04 April 2013
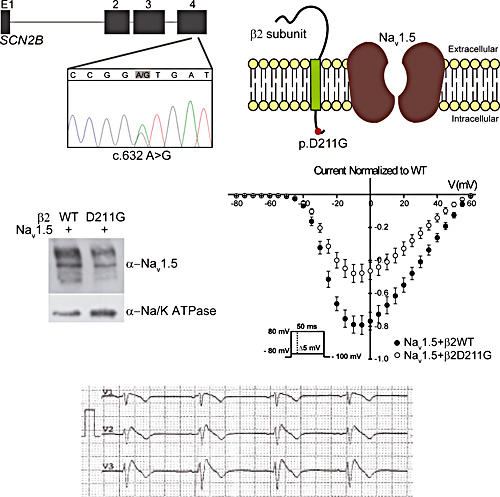
Brugada Syndrome (BrS) is a familial disease associated mainly to a loss-of-function of the cardiac sodium channel. In this work, we identified a novel missense mutation (p.Asp211Gly) in the sodium β2 subunit encoded by SCN2B, in a woman diagnosed with BrS. Our electrophysiological analysis showed that the β2D211G provoked a reduction in INa density by decreasing Nav1.5 cell surface expression. These results strongly suggest that SCN2B is a new candidate gene for BrS.
A New Coding System for Metabolic Disorders Demonstrates Gaps in the International Disease Classifications ICD-10 and SNOMED-CT, Which Can Be Barriers to Genotype–Phenotype Data Sharing
- Pages: 967-973
- First Published: 16 March 2013
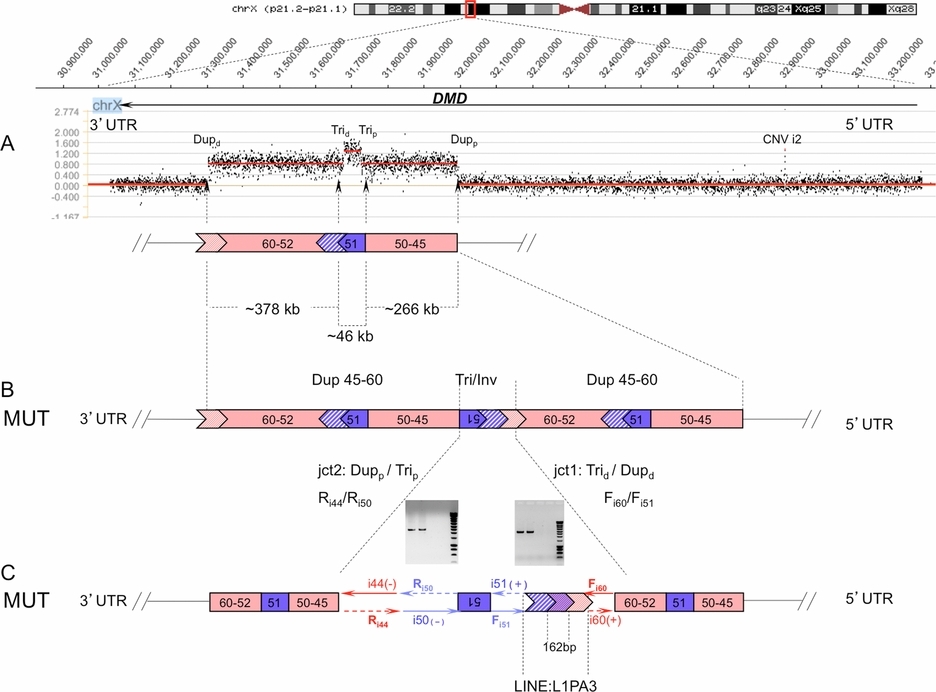
We report the molecular characterization of a complex duplication-triplication rearrangement involving exons 45–60 of the DMD gene which shares common genomic organization with the recently described duplication-inverted triplication-duplication (DUP-TRP/INV-DUP) events. Taking into consideration the presence of a predicted PRDM9 binding site in the near vicinity of the junction involving two inverted L1 elements and the inherent properties of X-Y chromosome recombination during male meiosis, we proposed an alternative two-step model for the generation of this X-linked DMD DUP-TRP/INV-DUP event.
Transduction-Specific ATLAS Reveals a Cohort of Highly Active L1 Retrotransposons in Human Populations
- Pages: 974-985
- First Published: 02 April 2013
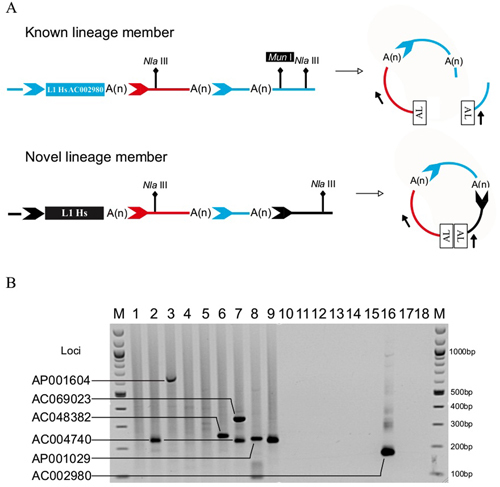
Long INterspersed Element-1 (L1) retrotransposons are the only independently mobile elements in the human genome. We developed Transduction-Specific Amplification Typing of L1 Active Subfamilies (TS-ATLAS), which utilizes L1-transduced genomic sequences, to identify a subset of highly active L1s genome-wide. TS-ATLAS enabled the characterization of the putative progenitor of an active disease-causing L1 lineage, and identified new retrotransposition events within two other “hot” L1 lineages.
RyR1 Deficiency in Congenital Myopathies Disrupts Excitation–Contraction Coupling
- Pages: 986-996
- First Published: 29 March 2013
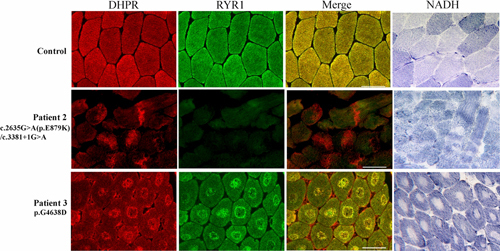
We provide evidence in this study that RyR1 deficiency caused by mutations in RYR1 gene is responsible for the collapse of excitation-contraction coupling by disrupting the physi-ological colocalization of the dihydropyridine receptor and ryanodine receptor in skeletal muscle. The upregulation of an alternative calcium regulating system via IP3R is identifi-ed in the condition of RyR1 deficiency and in-dicates the possible existence of a complex interplay on RyR1 and IP3R signalling cascade in the pathophysiology of RYR1-related conge-nital myopathies.
Mutations in the C-Terminal Domain of ColQ in Endplate Acetylcholinesterase Deficiency Compromise ColQ–MuSK Interaction
- Pages: 997-1004
- First Published: 29 March 2013
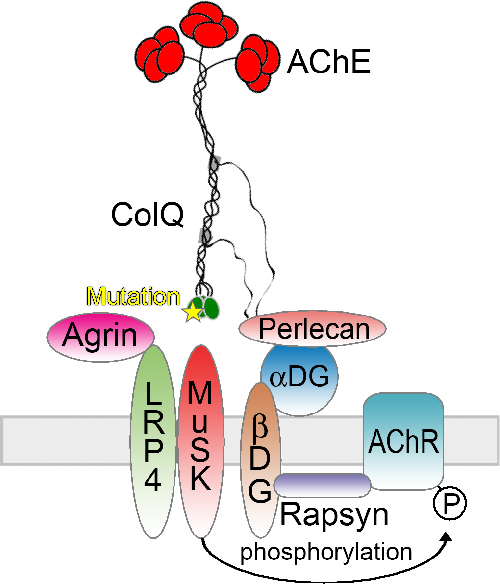
Missense mutations in COLQ encoding collagen Q (ColQ) compromise binding of ColQ to MuSK and lead to endplate acetylcholinesterase (AChE) deficiency. Defective binding has been proven by an in vitro overlay assay of mutant ColQ on skeletal muscle of Colq-/- mice and by an in vitro plate-binding assay on purified MuSK. We also confirmed that AAV8-mediated gene transfer of mutant ColQ into Colq-/- mice failed to express ColQ at the neuromuscular junction and to improve the motor deficits.
In-Depth Analysis of Hyaline Fibromatosis Syndrome Frameshift Mutations at the Same Site Reveal the Necessity of Personalized Therapy
- Pages: 1005-1017
- First Published: 29 March 2013
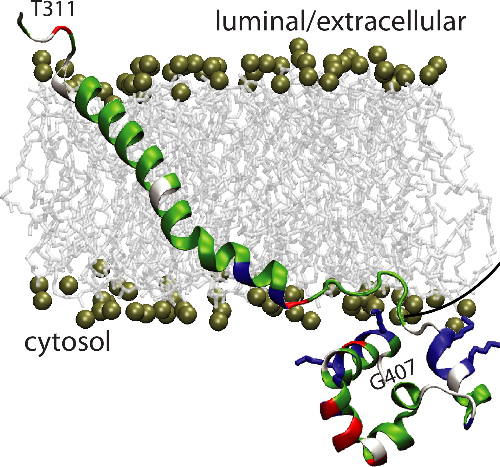
Hyaline Fibromatosis Syndrome is an autosomal recessive disease caused by mutations in ANTXR2. We show that targeting the non-sense mediated mRNA decay pathway leads to a rescue of ANTXR2 protein in patients carrying 1 base insertion but not in those carrying 2 base insertions at the same site. This highlights the importance of in depth analysis of the molecular consequences of specific patient mutations, which, even when they occur at the same site, can have drastically different consequences.
Relevance of Different Cellular Models in Determining the Effects of Mutations on SLC16A2/MCT8 Thyroid Hormone Transporter Function and Genotype–Phenotype Correlation
- Pages: 1018-1025
- First Published: 08 April 2013
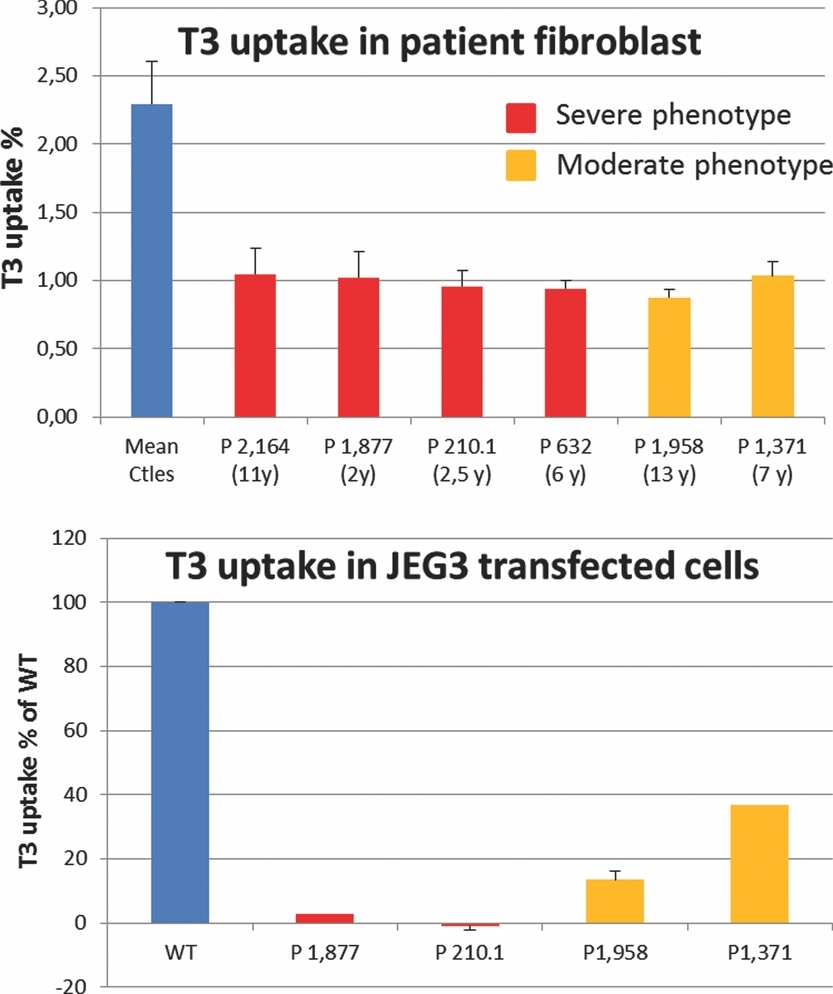
Demonstrating the deleterious effect of new sequence variants is critical for genetic diagnosis. In this paper, we demonstrate that functional studies measuring T3 transport in either patient fibroblasts or in JEG3 transfected cells are very easy and efficient tests in validating deleterious effects of new MCT8 mutations especially if there is still a doubt as to their pathogenicity. Moreover, our work establishes that JEG3 transfected cells are more relevant than fibroblasts in revealing a genotype–phenotype correlation for MCT8 diseases.
Exome Resequencing Identifies Potential Tumor-Suppressor Genes that Predispose to Colorectal Cancer
- Pages: 1026-1034
- First Published: 13 April 2013
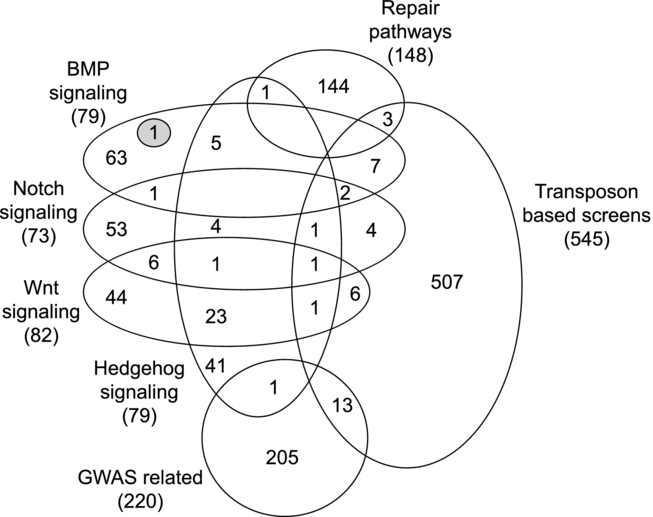
We exomeresequenced 50 sporadic patients with advanced CRC. To identify predisposition alleles, we sought rare/novel germline truncating mutations in 1138 genes considered likely to play a role in colorectal tumorigenesis. Thirty-two such mutations were identified, including an insertion in APC, thereby validating our approach. We sought somatic mutations in the corresponding genes in the CRCs of the patients harbouring the germline lesions and found biallelic inactivation of FANCM, LAMB4, PTCHD3, LAMC3 and TREX2, implicating their potential role in CRC-predisposition.
Targeted Next-Generation Sequencing can Replace Sanger Sequencing in Clinical Diagnostics
- Pages: 1035-1042
- First Published: 08 April 2013
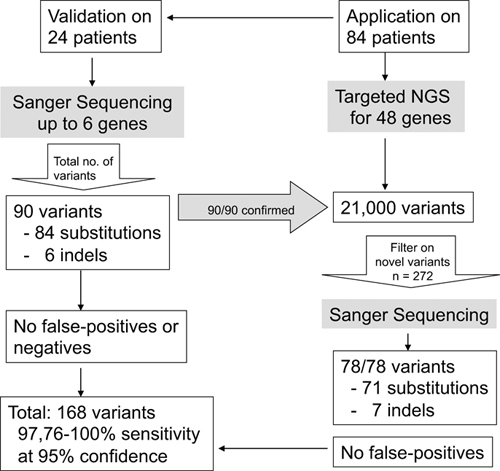
A NGS stand-alone diagnostic test for hereditary cardio-myopathies. Parallel analyses of 48 genes on a MiSeq identified ∼21,000 variants in 84 patients. The sensitivity and specificity of this test equals Sanger Sequencing. In total 168 variants were confirmed, no false-positives or false-negatives were detected.
How to Assess Causality of TMPRSS6 Mutations?
- Pages: 1043-1045
- First Published: 28 March 2013
The UBIAD1 Prenyltransferase Links Menaquinone-4 Synthesis to Cholesterol Metabolic Enzymes
- Page: 1046
- First Published: 30 April 2013
Allele-Specific Expression at the RET Locus in Blood and Gut Tissue of Individuals Carrying Risk Alleles for Hirschsprung Disease
- Page: 1047
- First Published: 08 May 2013