

Journal list menu

 Issue
IssueJournal of Computational Chemistry: Volume 38, Issue 31
i, 2665-2746December 5, 2017
Export Citations
Download PDFs
Cover Image
Cover Image, Volume 38, Issue 31
- Page: i
- First Published: 24 October 2017

Obtaining a material that can self-heal under ambient conditions is a great promise and challenge in smart materials science. This can be achieved by a special class of bonds called dynamic bonds. From these, diarylbibenzofuranone (DABBF) dynamic covalent bond is an appealing solution, but it cannot react with oxygen or it will not show self-healing behavior. On page 2675, G.R. Schleder, J.T. Arantes and A. Fazzio study the DABBF bond formation against oxidation using density functional theory (DFT). The cover image shows DABBF, oxygen, arylbenzofuranone (ABF), and its frontier orbitals. (DOI: 10.1002/jcc.24899)
Issue Information
Rapid Communication
Temperature-shuffled parallel cascade selection molecular dynamics accelerates the structural transitions of proteins
- Pages: 2671-2674
- First Published: 31 August 2017
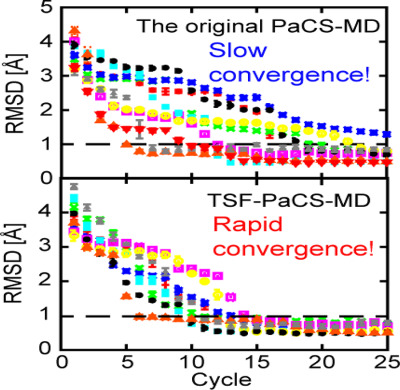
Parallel Cascade Selection Molecular Dynamics (PaCS-MD) is an enhanced conformational sampling method for searching structural transition pathways from a given reactant to a product. In the present study, Temperature-Shuffled PaCS-MD (TSF-PaCS-MD) is proposed as a further extension of PaCS-MD in which the temperatures are shuffled among different replicas at the beginning of each cycle of conformational resampling. Through evaluations, it was confirmed that TSF-PaCS-MD remarkably accelerated structural transitions of proteins than the original PaCS-MD.
Full Papers
Dynamic covalent bond from first principles: Diarylbibenzofuranone structural, electronic, and oxidation studies
- Pages: 2675-2679
- First Published: 27 July 2017
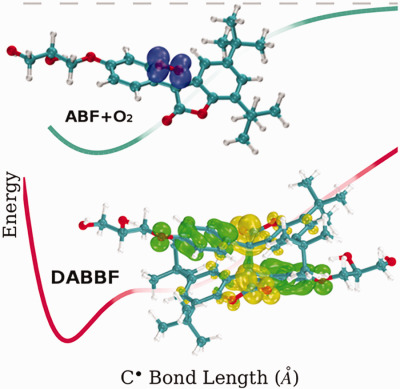
Obtaining a material that can self-heal under ambient conditions is a great promise and challenge in smart materials science. This can be achieved by a special class of bonds called dynamic bonds. From these, diarylbibenzofuranone (DABBF) dynamic covalent bond is an appealing solution, but it cannot react with oxygen or it will not show self-healing behavior. Here, using density functional theory, we study the DABBF bond formation against oxidation.
The influence of the metal cations and microhydration on the reaction trajectory of the N3 ↔ O2 thymine proton transfer: Quantum mechanical study
- Pages: 2680-2692
- First Published: 18 September 2017
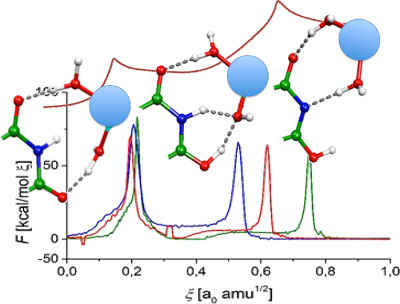
Intramolecular proton transfer (PT) process on thymine nucleobase between N3 and O2 atoms is explored in presence of hexacoordinated divalent metals cations Mg2+, Zn2+, and Hg2+. This PT proceeds as a two stages process. The first step involves the PT from one of the aqua ligands toward O2. The second stage is connected with the N3-proton abstraction. In the presence of the hexaaqua-cations, the activation barrier is at most 8 kcal/mol.
A computational study of samarium diiodide-induced cyclizations of N-oxoalkyl-substituted methyl indole-3-carboxylates—A rationale of the diastereoselectivity
- Pages: 2693-2700
- First Published: 02 September 2017
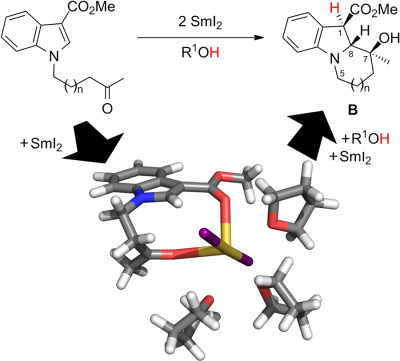
Samarium diiodide mediated reductive couplings of N-oxoalkyl-substituted methyl indole-3-carboxylates lead to benzannulated pyrrolizidines and indolizidines with a surprisingly high diastereoselectivity. Gibbs energies of reaction steps are calculated with dispersion corrected DFT and the solvation model D-COSMO-RS. In contrast to previous mechanistic proposals, the diastereoselectivity in the cyclization is found to be due to the formation of an energetically highly favorable chelate complex in which the final relative configuration is already preformed.
Investigation of challenging spin systems using Monte Carlo configuration interaction and the density matrix renormalization group
- Pages: 2701-2712
- First Published: 02 September 2017
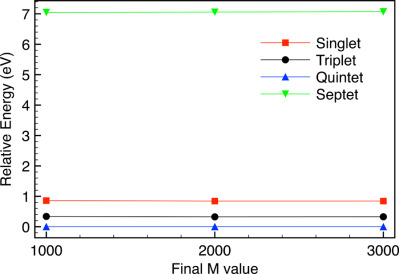
Challenging spin systems can often have multireference character that requires computationally intensive methods and significant decisions by a user to achieve accurate results. We investigate if Monte Carlo configuration interaction and the density matrix renormalization group can be applied in an approach that moves towards a “black box” implementation. We generally find qualitative agreement for the ordering of spin states for the systems considered.
Comparison of a quantum random number generator with pseudorandom number generators for their use in molecular Monte Carlo simulations
- Pages: 2713-2720
- First Published: 18 September 2017
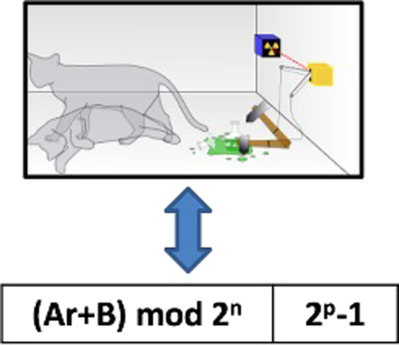
Four pseudorandom number generators were compared with a physical, quantum-based random number generator using the NIST suite of statistical tests. We then measured the effect of the five random number generators on various calculated properties in different Markov-chain Monte Carlo simulations. The results show that poor quality pseudorandom number generators produce results that deviate significantly from those obtained with the quantum-based random number generator. In contrast, the Mersenne Twister pseudorandom generator and a 64-bit Linear Congruential Generator with a scrambler produce results that are statistically indistinguishable from those obtained with the quantum-based random number generator.
Adaptive-numerical-bias metadynamics
- Pages: 2721-2729
- First Published: 26 September 2017
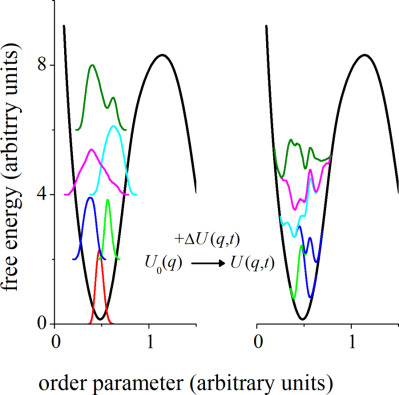
Four pseudorandom number generators were compared with a physical, quantum-based random number generator using the NIST suite of statistical tests. We then measured the effect of the five random number generators on various calculated properties in different Markov-chain Monte Carlo simulations. The results show that poor quality pseudorandom number generators produce results that deviate significantly from those obtained with the quantum-based random number generator. In contrast, the Mersenne-Twister pseudorandom generator and a 64-bit Linear Congruential Generator with a scrambler produce results that are statistically indistinguishable from those obtained with the quantum-based random number generator.
Ergodicity and model quality in template-restrained canonical and temperature/Hamiltonian replica exchange coarse-grained molecular dynamics simulations of proteins
- Pages: 2730-2746
- First Published: 22 September 2017
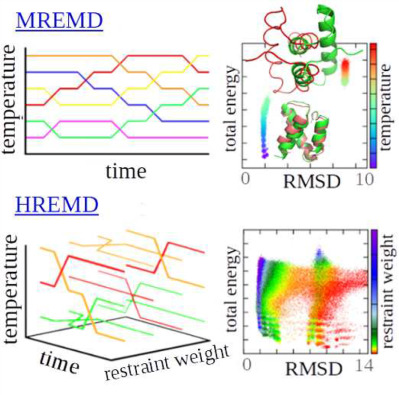
Illustration of the difference between restrained temperature multiplexed replica exchange molecular dynamics (MREMD, top) and restrained Hamiltonian multiplexed replica exchange molecular dynamics (HREMD, bottom). Exchange of temperatures only (MREMD) can result in getting trapped in topological mirror image structures (top right), while adding exchanges of the restraint weights (HREMD) enables the system to escape from such structures, owing to the weakening or removing restraints in a part of a trajectory (bottom right).
Sign up for email alerts
Tools
