


All Ecology and Evolution Articles
Export Citations
Download PDFs
-
The Mitochondrial Genome of the Imperiled Goliath Grouper Epinephelus itajara: Selective Pressures in Protein Coding Genes, Secondary Structure of tRNA Genes, and Phylogenetic Placementoa
Graphical Abstract
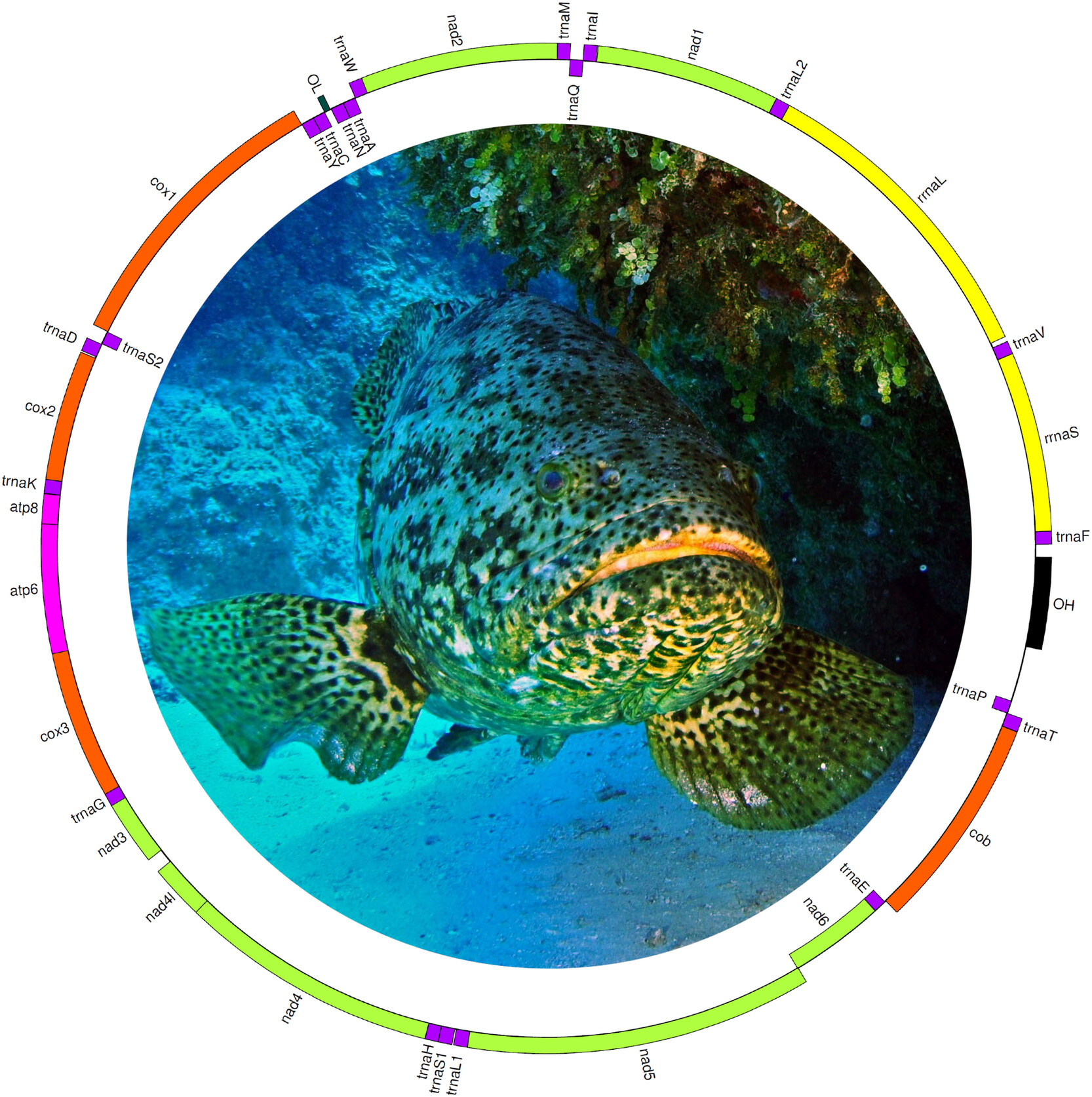
This study presents the complete mitochondrial genome of the critically endangered Goliath Grouper (Epinephelus itajara), totaling 16,561 bp and comprising 13 protein-coding genes, 22 tRNAs, and two rRNAs. Genomic analysis revealed conserved gene order, A + T-rich codon usage, and purifying selection across all protein-coding genes, providing valuable resources for conservation genetics and species identification. Photograph by Albert Kok, used with permission.
-
Overwintering and Resident Birds in Qatar: Explorations With DNA Barcodingoa
Graphical Abstract
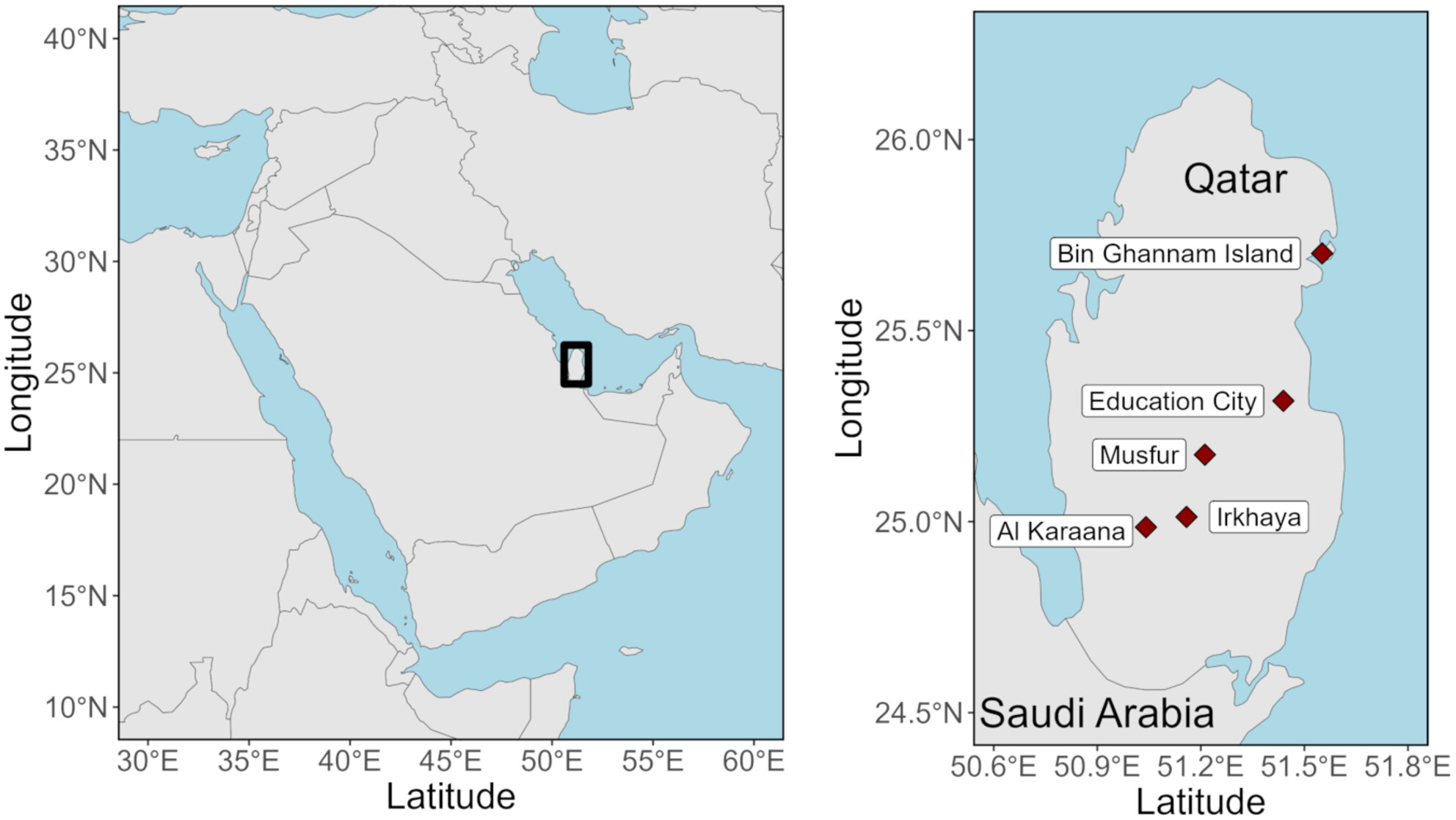
We obtained DNA barcodes for 115 birds (34 species, 12 orders) in Qatar, a country with no previous barcode sequence data for wild birds. We provide insights on locally breeding species and overwintering migrants.
-
Complete Mitochondrial Genome and Its Phylogenetic Analysis of Oides decempunctatus (Coleoptera: Chrysomelidae)oa
Graphical Abstract
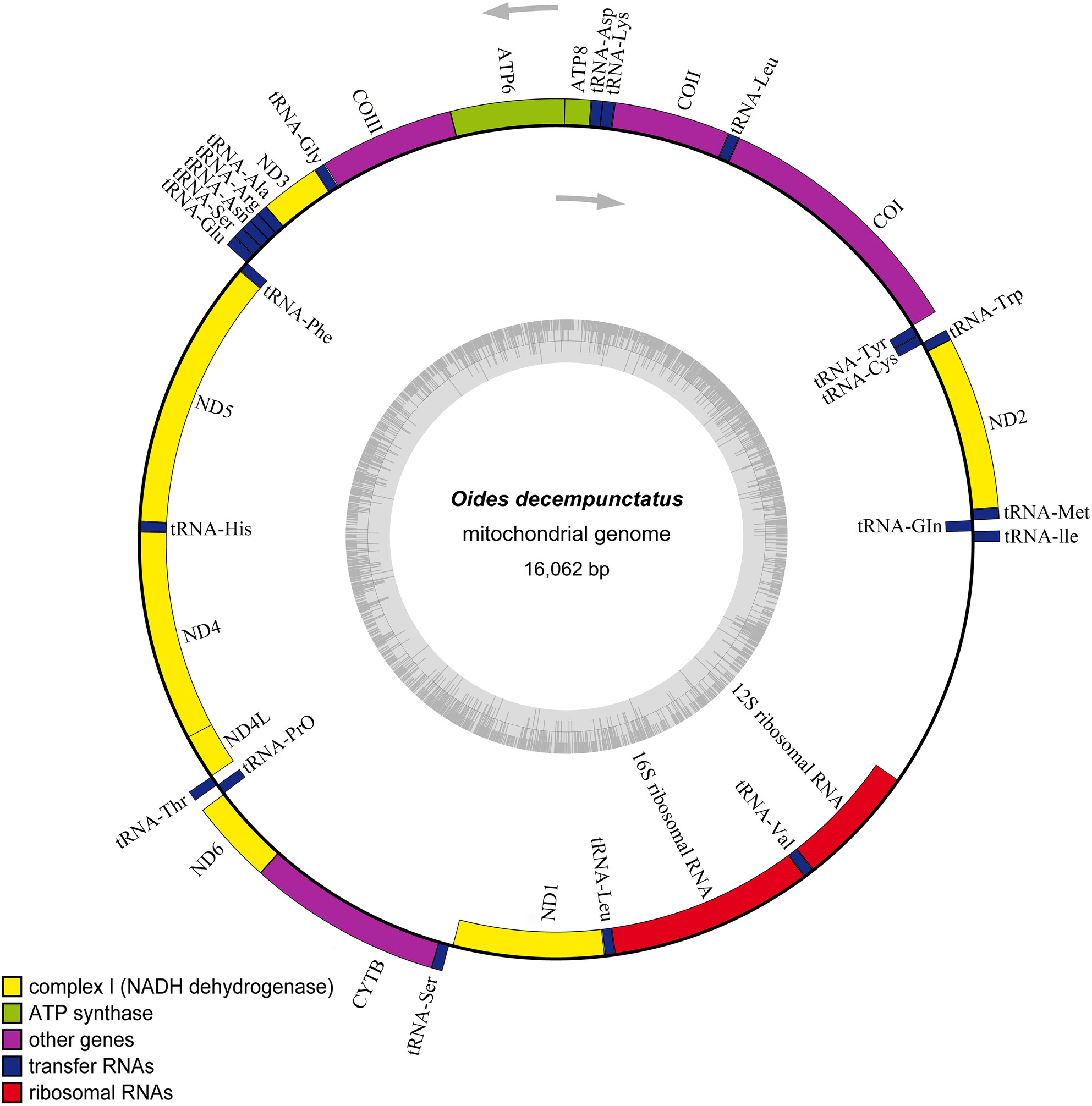
This study provides important foundational information for further research on the mitochondrial genomes of Chrysomelidae.
-
Genotyping by Amplicon Sequencing (GBAS) With Newly Developed SSR and EPIC Markers Reveals Structure in Populations of the Green Toad (Bufotes viridis) Across Rural and Urban Environmentsoa
Graphical Abstract

We developed and tested SSR and EPIC markers for the green toad (Bufotes viridis), demonstrating their effectiveness using genotyping by high-throughput amplicon sequencing (GBAS) across urban and rural populations. EPIC markers exhibited fewer null alleles and provided more ecologically coherent clustering results, while SSRs reflected drift-induced patterns. Our findings indicate that B. viridis may retain genetic diversity in urban environments and highlight the value of combining EPIC and SSR markers, particularly in fragmented habitats prone to genetic drift.
-
Genomic Phylogenetic Analysis of Physaliastrum and Archiphysalis (Solanaceae): Insights From Chloroplast Genomes Indicate Distinct Evolutionary Relationshipsoa
Graphical Abstract

This research involved the sequencing and analysis of chloroplast genomes from nine species across four genera of the Solanaceae family, specifically two species of Archiphysalis, five species of Physaliastrum, and the taxa Tuberowithania pengiana and Tubocapsicum anomalum. We conducted a phylogenetic analysis that clarifies the relationships among these genera and confirms their distinct generic statuses. Additionally, this study provides genomic evidence supporting their classification and highlights the close relationship between A. sinensis and A. chamaesarachoides.
-
Comparative and Phylogenetic Analyses of Mitochondrial Genomes in Carabidae (Coleoptera: Adephaga)oa
Graphical Abstract
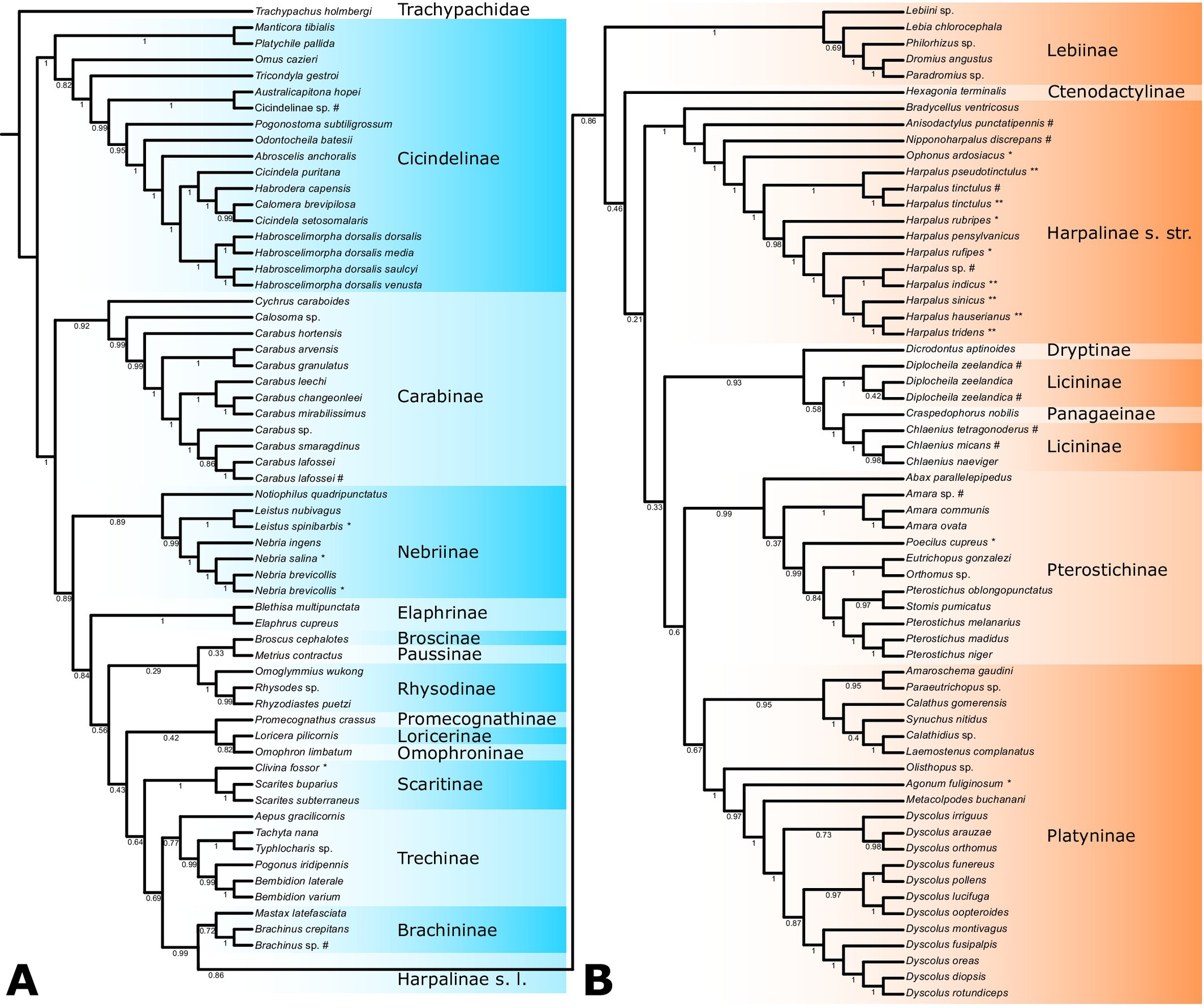
In this study, six complete mitochondrial genomes (mitogenomes) from the genus Harpalus are reported. Additionally, 13 cases of suspected misidentifications in public Carabidae mitogenomes from GenBank were identified, with potential corrections suggested. Bayesian inference and maximum likelihood approaches were used to infer the phylogenetic relationships within Carabidae based on 121 mitogenomes and various datasets.
-
Insight Into the Nuclear and Mitochondrial Genome of the Caribbean King Crab Maguimithrax spinosissimus (Crustacea: Brachyura: Mithracidae) to Support Fisheries Management and Conservation Initiativesoa
Graphical Abstract
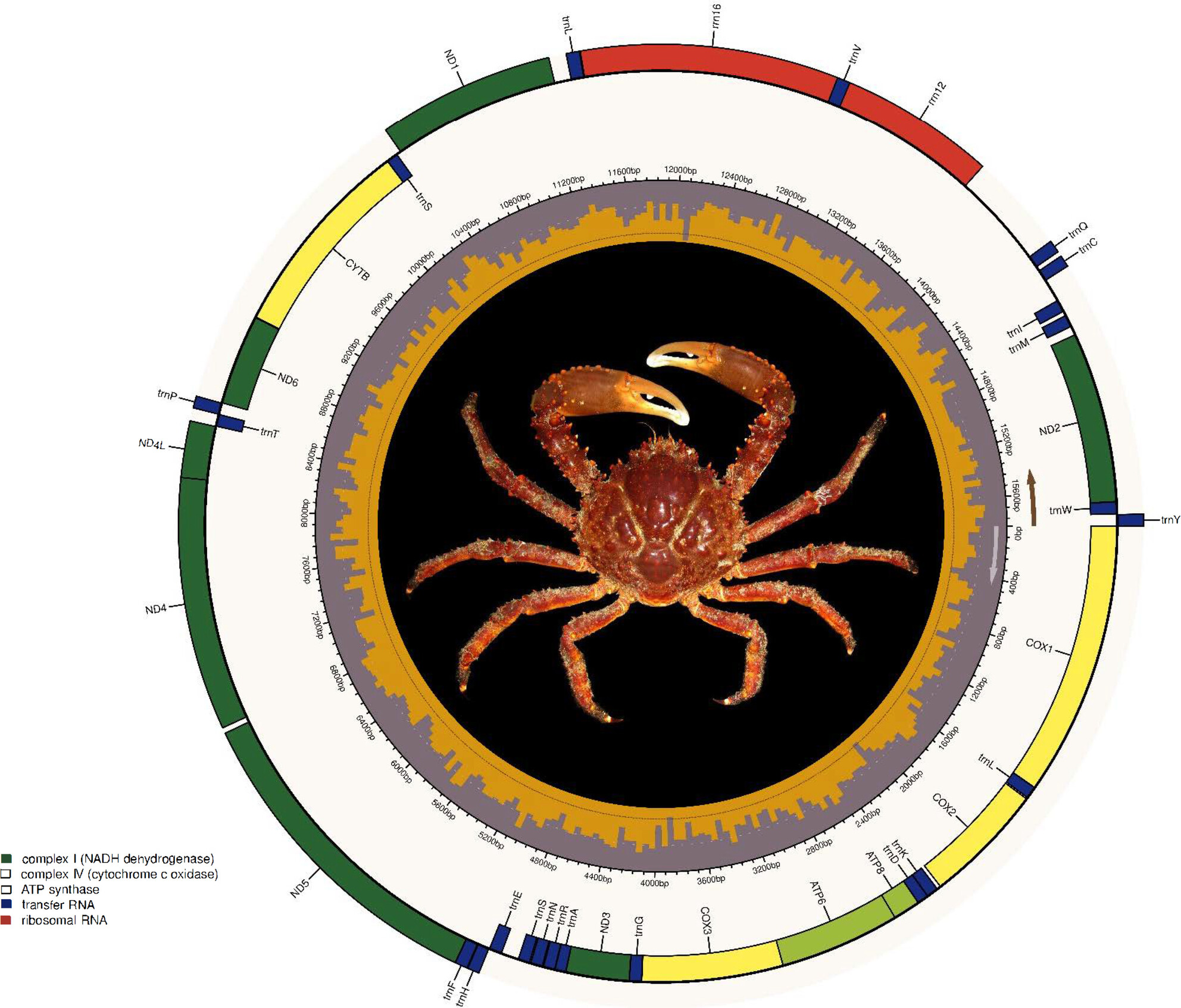
Genomic resources for the Caribbean King Crab.
-
Determining Zygosity With Multiplex Kompetitive Allele-Specific PCR (mxKASP) Genotypingoa
Graphical Abstract
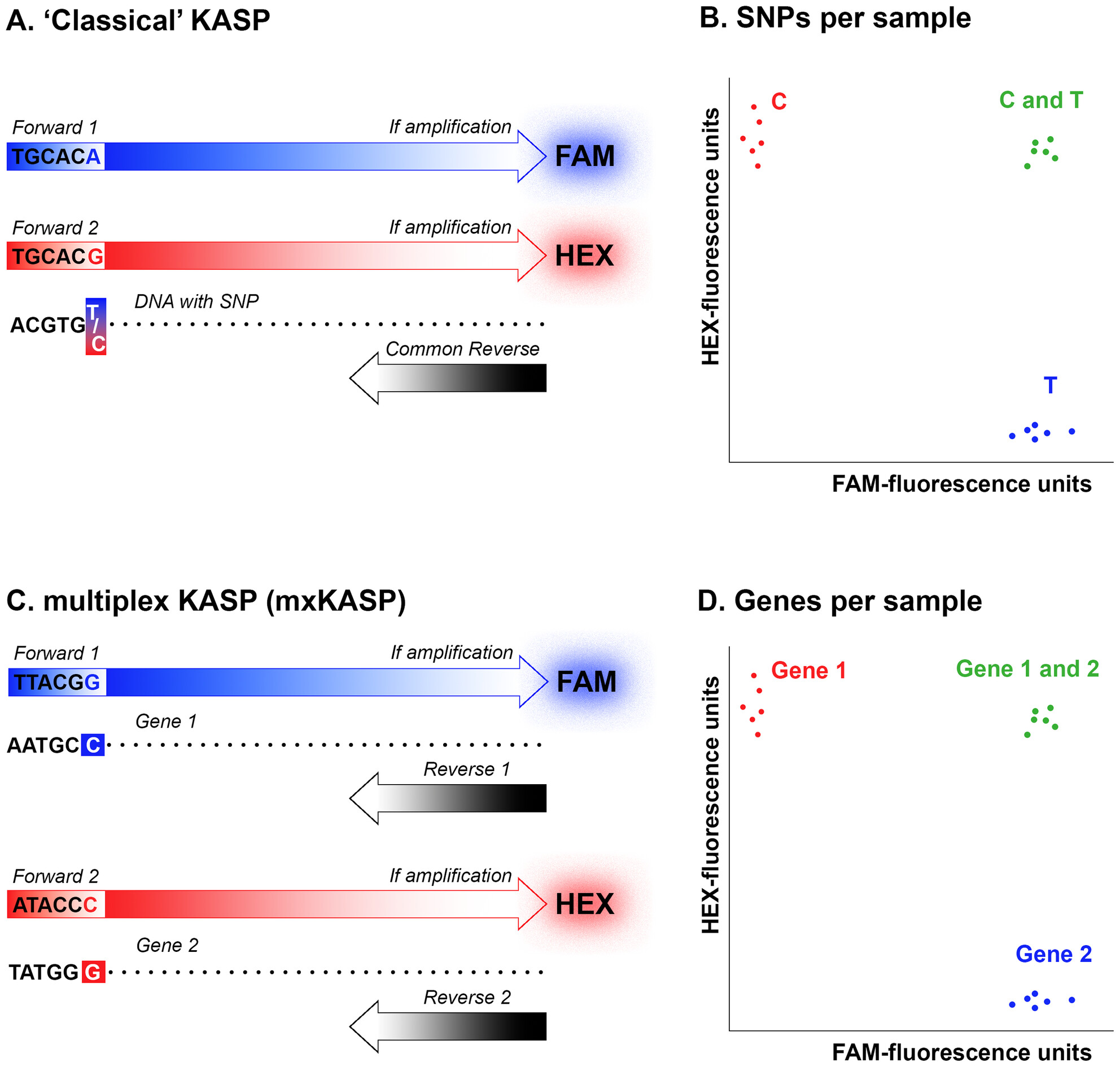
Our study introduces mxKASP, a modified Kompetitive Allele-Specific PCR technique that allows for determination of zygosity in diploid organisms by targeting two entirely different, non-homologous markers. This is different from traditional KASP, in which a single marker is targeted, but with two different, SNP-specific primers.
-
Unfolding the Complete Chloroplast Genome of Myrica esculenta Buch.-Ham. ex D.Don (1825): Advancing Phylogenetic Insights Within Fagales and Pioneering DNA Barcodes for Precise Species Identificationoa
Graphical Abstract
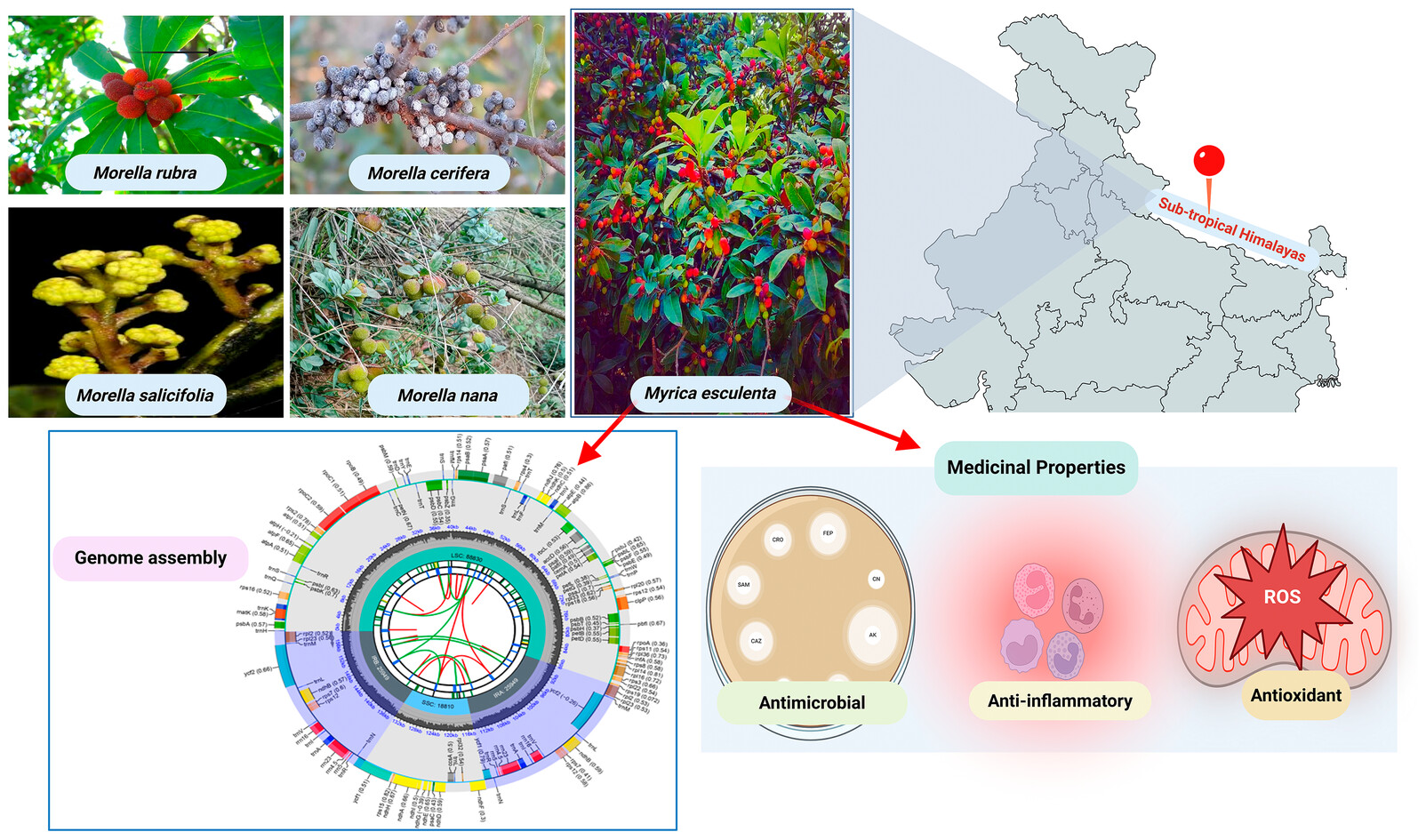
This study presents the complete chloroplast genome of Myrica esculenta (159,538 bp) and its phylogenetic placement within the Fagales order. Phylogenetic analysis using the ycf1 gene confirmed a monophyletic clade with other Morella species, highlighting ycf1 as a promising DNA barcode for species identification. The findings enhance phylogenetic insights and support applications in plant breeding, herbal drug authentication, and evolutionary studies.
-
The Complete Chloroplast Genome Sequence of Callisia fragrans (Lindl.) Woodson (Commelinaceae)oa
Graphical Abstract
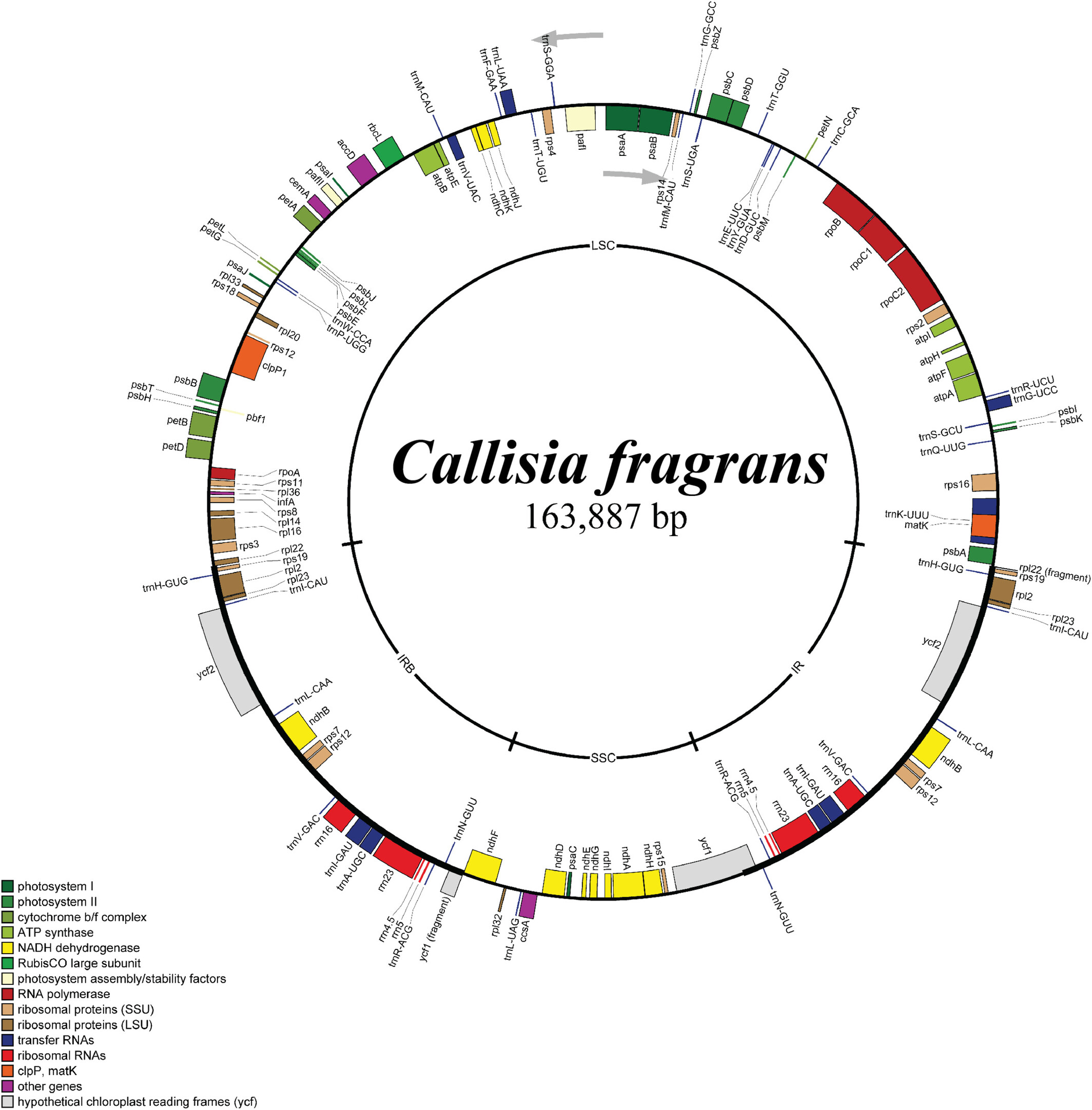
This research reported the complete chloroplast genome of Callisia fragrans, an ornamental and medicinal plant in the Commelinaceae family. The comparative genomic analysis revealed a unique feature of the C. fragrans chloroplast genome within the Callisia genus.
-
Genetic Diversity of Chinese Giant Salamanders Under the Context of Translocation Using Novel Development of Genomic SSR Markersoa
Graphical Abstract
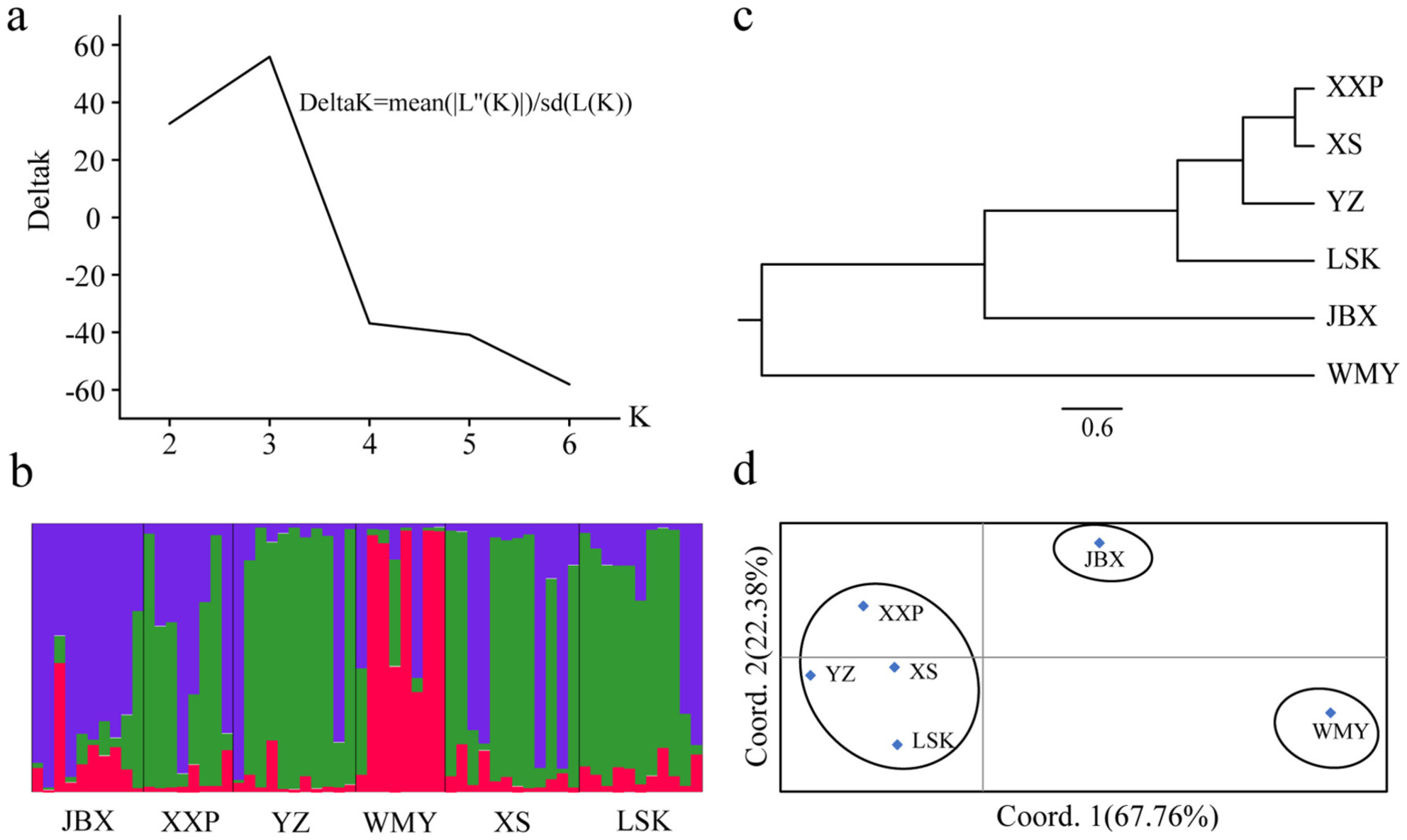
This study developed 19 novel genomic SSR markers for the critically endangered Chinese giant salamander (Andrias spp.) using next-generation sequencing, revealing 316,313 SSR loci. Analysis of 60 wild individuals across six sites in Hunan Zhangjiajie Giant Salamander National Nature Reserve identified the highest genetic diversity in the long-term translocated Jinbianxi population and the lowest in the natural Wumuyu site, with significant differentiation between natural populations compared to recently reintroduced groups. The findings emphasize the need for genetic monitoring to mitigate hybridization risks and improve conservation strategies for this imperiled species.
-
Phylogenetic Variation of Periophthalmus Species Living in the Mekong Delta, Vietnam, Using Cytb Sequenceoa
Graphical Abstract

This study investigates the genetic relationships of three Periophthalmus mudskipper species (P. chrysospilos, P. gracilis, and P. variabilis) from the Mekong Delta using mitochondrial cytochrome b (Cytb) gene analysis. The findings reveal significant interspecific genetic divergence (15%–18%) and distinct phylogenetic sub-clades supported by morphological differences and high bootstrap values. These results provide valuable insights into the evolutionary history and genetic diversity of amphibious fish in this region.
-
Eight Novel Microsatellite Loci in a Declining Grassland Songbird, the Bobolink (Dolichonyx oryzivorus)oa
Graphical Abstract

This paper introduces ten new polymorphic microsatellite loci for genetic analysis of Bobolinks (Dolichonyx oryzivorus, Family Icteridae), which, like many grassland songbirds, are declining across their native range in North and South America. The informativeness of these hypervariable loci was confirmed based on the genotyping of 152 adult individuals from a well-studied population in Shelburne, Vermont, USA. These loci will provide an important tool for assessing relatedness between individuals and monitoring population dynamics over time.
-
The Complete Mitochondrial Genome of Squalus cubensis and Comparative Mitogenomics and Phylomitogenomics of the Family Squalidaeoa
Graphical Abstract
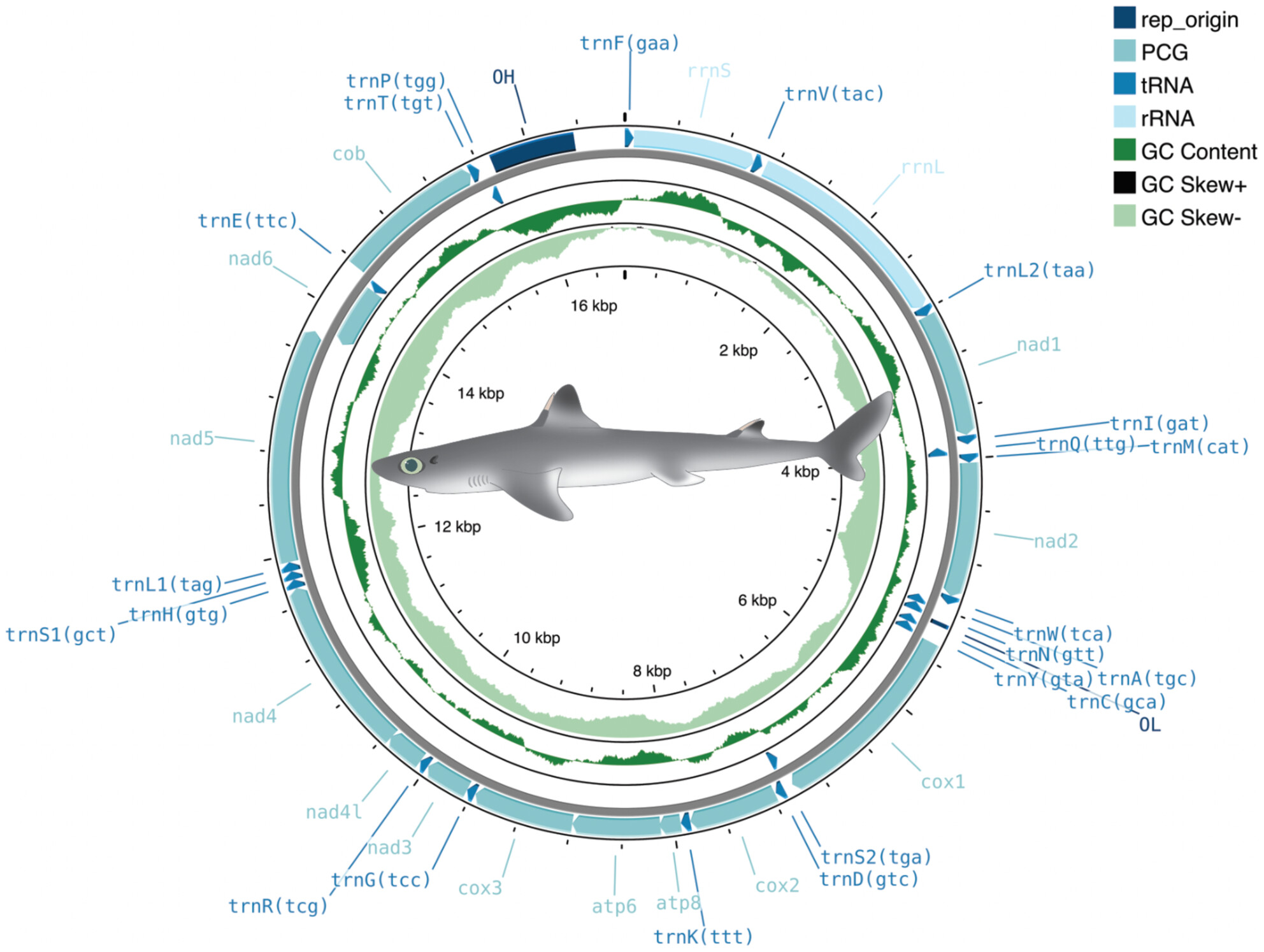
The complete mitochondrial genome of Squalus cubensis.
-
Synteny Enabled Upgrade of the Galapagos Giant Tortoise Genome Improves Inferences of Runs of Homozygosityoa
Graphical Abstract
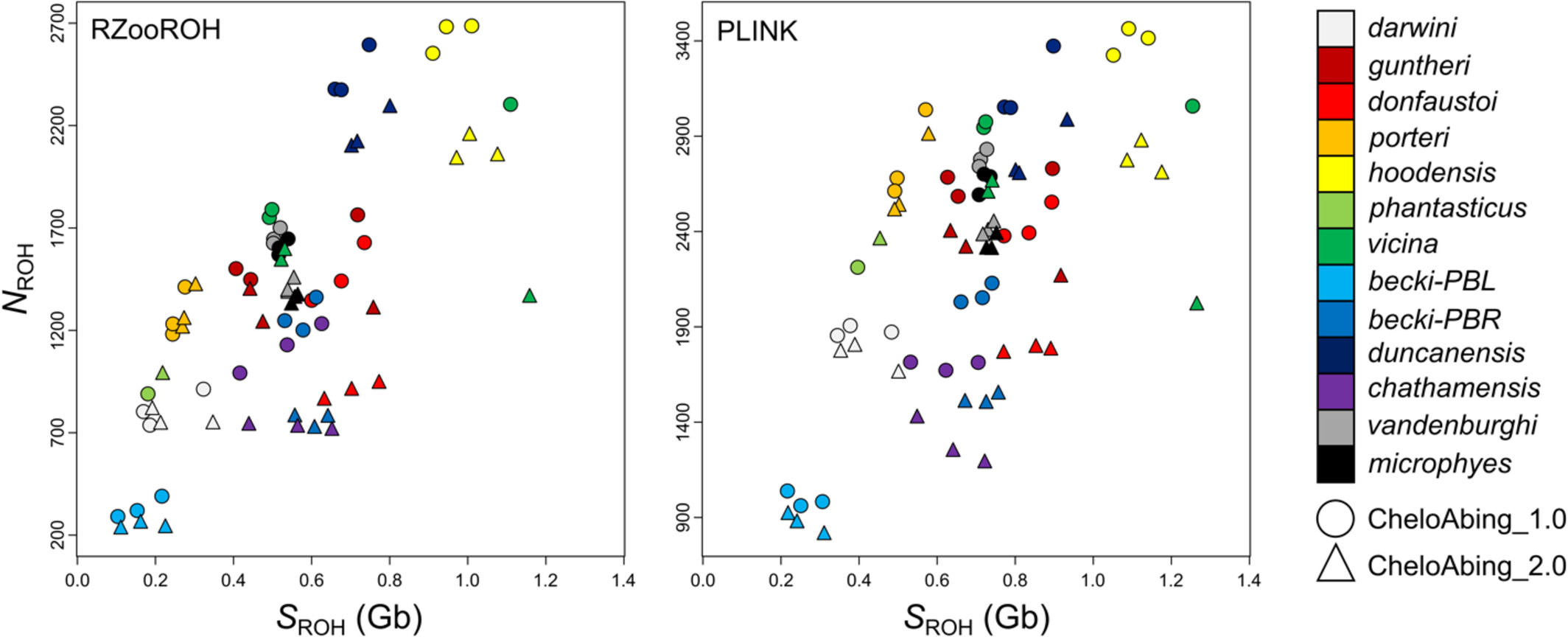
Here we update the reference genome for the Pinta Island Galapagos giant tortoise (Chelonoidis abingdonii) through rescaffolding against the most closely related chromosome-level genome assembly, the Aldabra giant tortoise (Aldabrachelys gigantea). This effort resulted in a much more contiguous genome, CheloAbing_2.0, with an N50 that is two orders of magnitude longer, which allowed us to test the mechanisms by which a fragmented assembly may over- or underestimate the number and extent of ROH. Analysis of resequencing data of 13 individuals resulted in individual estimates of inbreeding, including ROH proportion (FROH), number (NROH), and cumulative length (SROH), that were statistically different from those derived from the earlier genome assembly.
-
SNP-RFLP Markers for the Study of Arabidopsis lyrataoa
Graphical Abstract
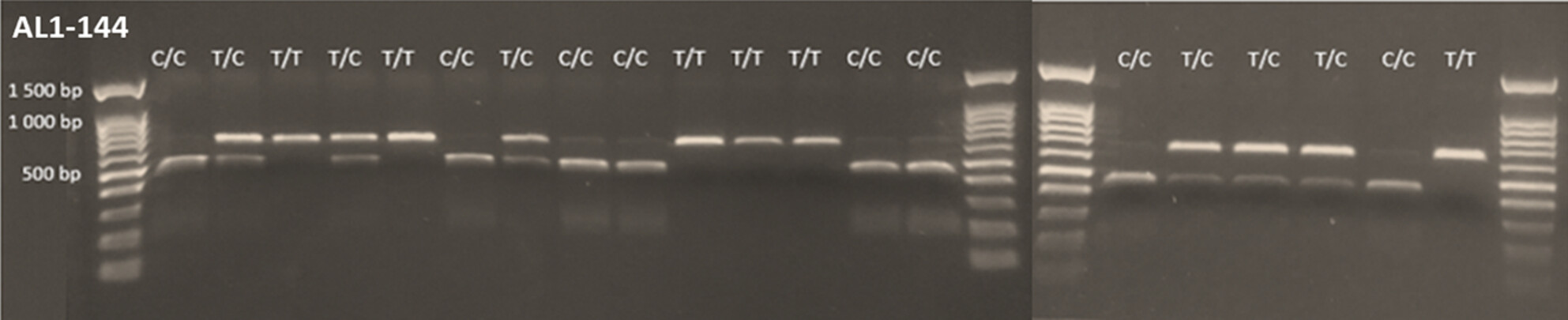
We outline the development of highly variable, easy-to-use, and cost-effective genetic markers for the analysis of Arabidopsis lyrata. These markers may be useful for the further development of this plant as a model for the study of micro-evolutionary processes, including evolutionary modifications to plant mating systems.
-
The Complete Chloroplast Genome of Tornillo (Cedrelinga cateniformis Ducke 1922, Fabaceae)oa
Graphical Abstract
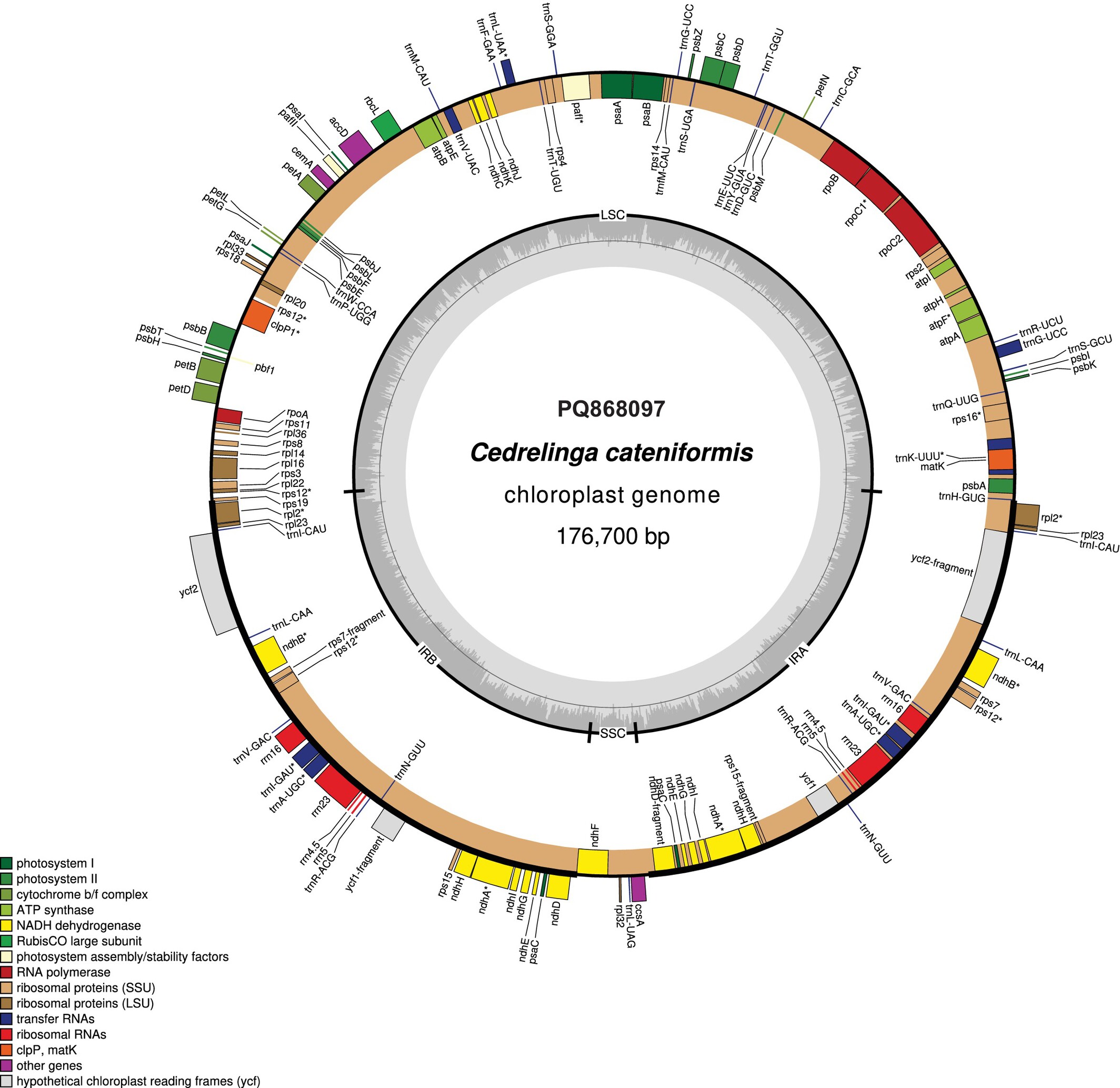
Tornillo (Cedrelinga cateniformis) is a tropical tree from the Fabaceae family, valued as a substitute for overexploited timber species. This study presents the first complete chloroplast genome sequencing of Tornillo, revealing a 176,700 bp structure with 138 genes and confirming its phylogenetic placement within the Ingeae tribe.
-
Mitochondrial Genomes of Three Melampus Species (Ellobiidae; Gastropoda)oa
Graphical Abstract
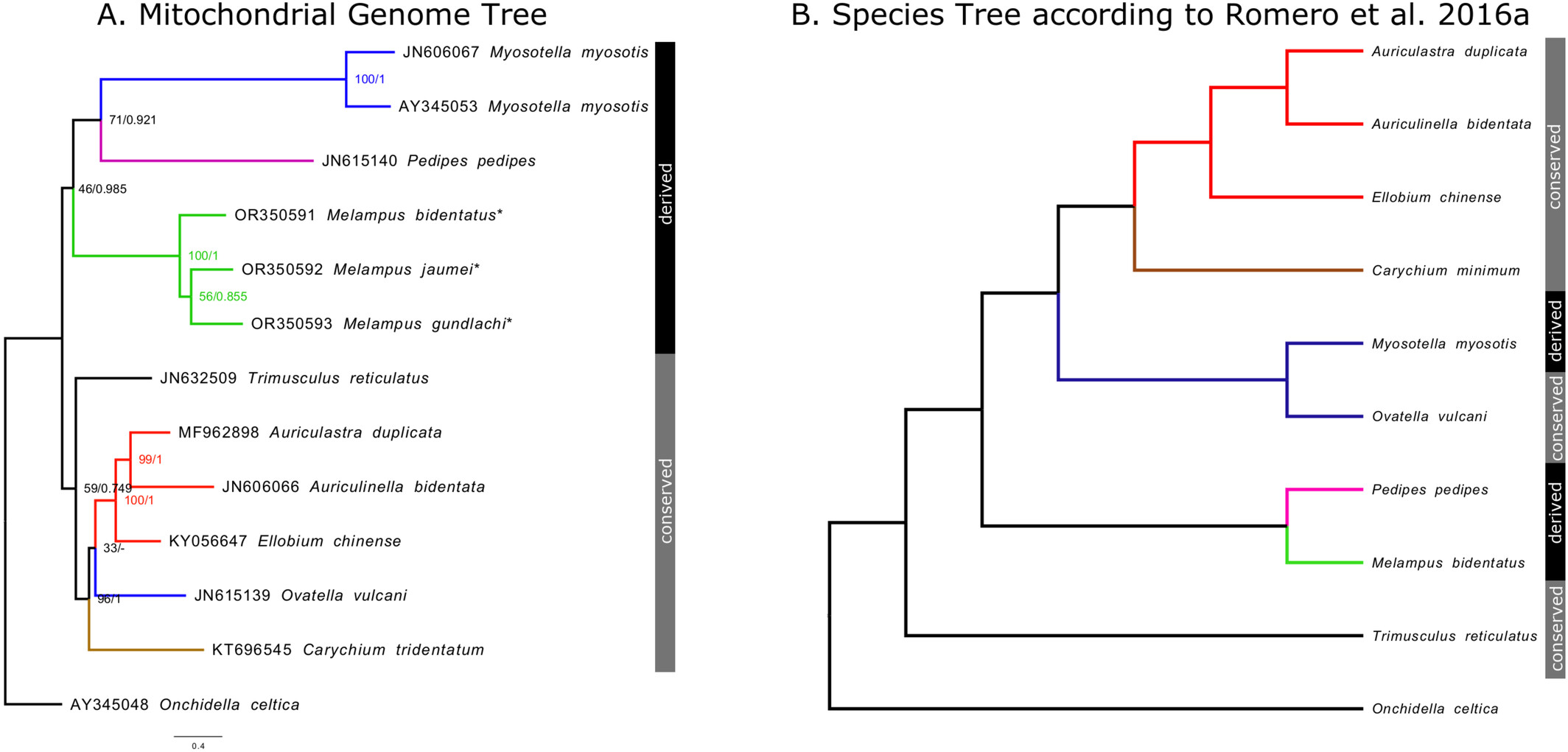
The mitochondrial genomes of three gastropod species from the family Ellobiidae—Melampus bidentatus, Melampus jaumei, and Melampus gundlachi—were assembled and annotated and found to have a gene order that diverges from the conserved gene order that is recovered in most genera within Ellobiidae and—more generally—within Euthyneura. Mitochondrial genomes within Ellobiidae that differ from the conserved gene order were found to be more divergent compared to each other than the mitochondrial genomes that have the conserved gene order, indicating variable evolutionary rates of mitochondrial genes within the family. Comparisons among 130 mitochondrial genomes within Euthyneura supported the presence of a control region preceding the isoleucine-tRNA, including in the novel genomes presented here.
-
Complete Mitochondrial Genome Analysis Reveals Genetic Diversity in the Narrow–Ridged Finless Porpoise (Neophocaena asiaeorientalis) Across East Asian Watersoa
Graphical Abstract
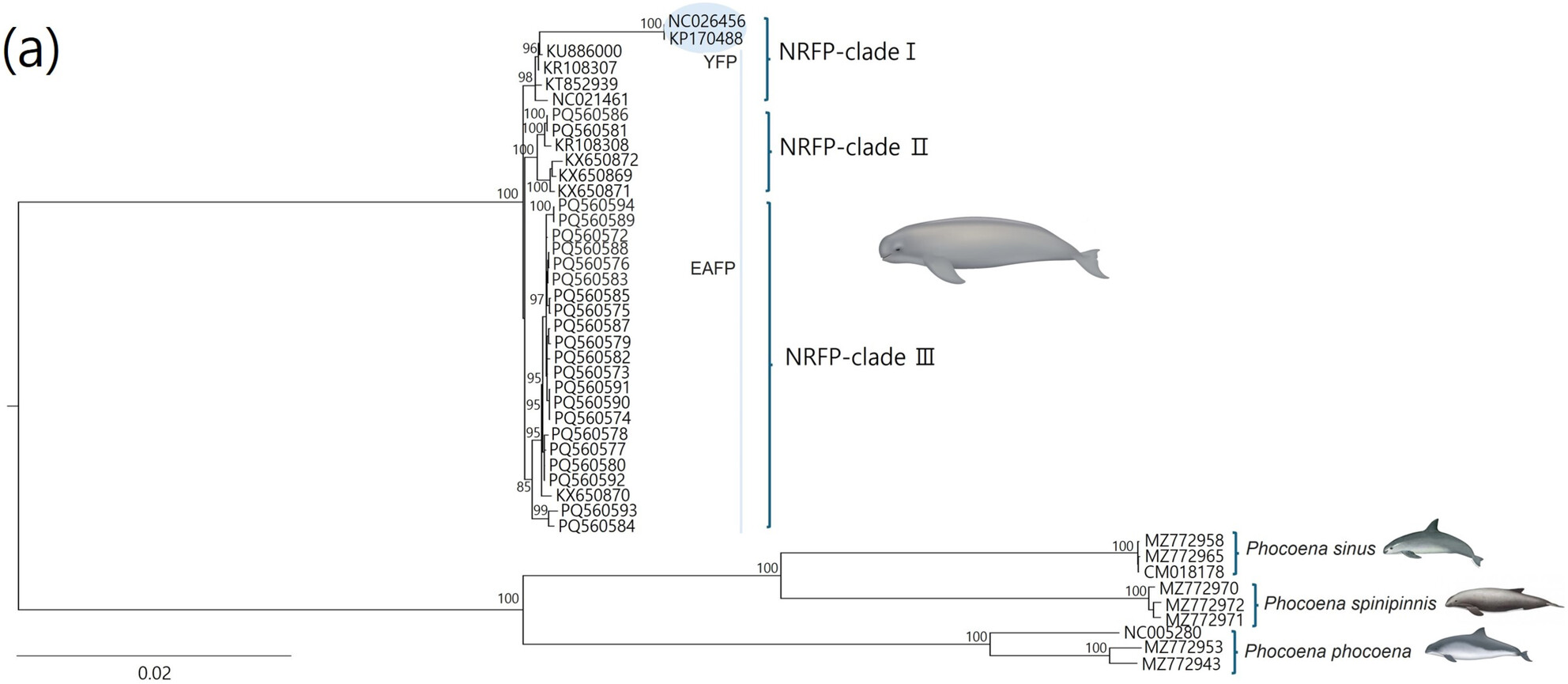
The narrow-ridged finless porpoise (Neophocaena asiaeorientalis Pilleri & Gihr, 1972) is one of the most endangered cetacean species inhabiting East Asian waters. Complete mitogenome analysis offers accurate phylogenetic insights; however, complete mitogenome sequences for the narrow-ridged finless porpoise have so far been restricted to specific regions, mainly in China, and no sequences are available from Korean or Japanese populations. To address this gap, in this study, we developed a multiplex PCR primer panel to sequence the complete mitochondrial genome of the porpoise and sequenced 23 individuals of N. a. sunameri, a subspecies of N. asiaeorientalis, from Korean waters using next-generation sequencing. Phylogenetic analyses based on maximum likelihood and Bayesian inference revealed three major, well-supported monophyletic clades within the species. Two sequences of the Yangtze finless porpoise (N. a. asiaeorientalis), another subspecies, displayed significantly higher genetic divergence compared to N. a. sunameri sequences. The 23 mitochondrial genome sequences exhibited a nucleotide diversity of 0.142% and a haplotype diversity of 99.6%, with 22 unique haplotypes identified. These findings contribute to our understanding of the evolutionary history and genetic diversity of the species, providing valuable insights for future conservation efforts and further genetic research.