

Phosphatase Dysregulation in Cancer: Signaling Pathways and Therapeutic Opportunities
ABSTRACT
Phosphatases are increasingly recognized as critical regulators of cancer biology, with important roles in both tumor cells and the tumor immune microenvironment (TIME). These enzymes modulate intracellular signaling pathways that control tumor growth, immune evasion, and metastasis. Although phosphatases were once considered undruggable, recent advances have highlighted their therapeutic potential. Despite growing evidence, phosphatases remain underexplored as drug targets, with no approved therapies to date. This review presents an in-depth overview of phosphatase classification based on catalytic domain similarities and explores their diverse functions as tumor suppressors, oncogenic drivers, or context-dependent regulators. We describe how phosphatases such as PTPN6, PTPN22, and DUSPs regulate key pathways like RAS/MAPK and PI3K/AKT in both tumor and immune cells. Additionally, we discuss the role of phosphatases in shaping the tumor microenvironment through exosome secretion. This review highlights current therapeutic strategies, including small molecules and antibodies, and their synergistic effects with kinase inhibitors and immune checkpoint blockade. By summarizing recent advances, this paper underscores the need for deeper mechanistic insights into phosphatase function in cancer and immunity. Understanding these mechanisms will be key to unlocking their potential as novel therapeutic targets in oncology.
Graphical Abstract
Schematic depiction of the role of phosphatases in cancer, highlighting their involvement in proliferation, exosome secretion, and immunosuppression. The SHP2 crystal structure (PDB:5EHR) is shown as the representative phosphatase.

1 Introduction
Protein modification is a dynamic process regulated by the fine balance between protein tyrosine kinase (PTKs) and protein tyrosine phosphatase (PTPs) activity. This balance dictates overall protein function and cellular homeostasis. Dysregulated PTK and PTP activities are a hallmark of many malignancies, such as breast, pancreatic, and lung cancers [1]. While PTKs have been extensively studied and targeted with therapeutics, PTPs have emerged as promising but underexplored targets for cancer therapy [2, 3]. PTPs exhibit diverse roles in the cellular environment, functioning as tumor suppressors, positive regulators of oncogenic pathways, or both. They modulate key cancer hallmarks such as immunosuppression in the tumor microenvironment and metastasis by interacting with various substrates in the cell [3, 4]. Tyrosine kinase inhibitors (TKIs) resistance arises from the overactivation or loss of phosphatase activity and drives tumor progression and carcinogenesis through activating mutations or other mechanisms. Thus, targeting PTPs offers an untapped therapeutic avenue in cancer research, as seen by the robust antitumor activity of targeting phosphatase In Vitro and vivo cancer models [5-7].
Several phosphatases (i.e., PTPN22) are ubiquitously expressed in immune cells, where they play pivotal roles in regulating immune cell activation [3]. They modulate pathways downstream of the T-cell receptor or immune checkpoint inhibitors such as PD-1 and CTLA-4, making them attractive targets for immuno-oncology [8, 9]. Additionally, phosphatases regulate effector T-cell differentiation, myeloid cell differentiation, and antigen presentation, underscoring their critical roles in the immune tumor microenvironment [3, 10]. Phosphatase dysregulation in cancer cells has a wide range of implications, including uncontrolled proliferation, angiogenesis, and metastasis. This dysregulation primarily occurs through the phosphatase's activity in the RAS/MAPK, PI3K/AKT, and Hippo oncogenic signaling pathways [11-13].
The role of phosphatases depends on their substrates and the specific cell type in which they are expressed. Whether in tumor cells or non-tumoral cells (e.g., immune cells), their function in cancer is dictated by the cellular context [14]. Phosphatases are classified based on catalytic domain similarity and can promote, suppress, or play dual roles in tumor growth, depending on their biological function and cellular context [3, 15]. PTPN22 and DUSP1/6 are known to promote tumorigenesis, while SHP1 primarily functions as a tumor suppressor [16-19]. However, PTPN2, PP2A, PTP1B, and SHP2 exhibit dual roles, acting as either tumor promoters or suppressors depending on the specific cellular context [20-26]. However, phosphatase dysregulation is not limited to the cellular level; it also affects cell-to-cell communication through exosome secretion [27-30]. Phosphatase dysregulation significantly suppresses tumor-immune cell crosstalk, reducing immune cell activation and overall inflammation in the tumor microenvironment (TME) through exosome secretion, thereby contributing to immunotherapy resistance [28, 29, 31-33]. Thus, phosphatases play a crucial role in tumor progression through intrinsic (cellular) mechanisms, such as activating the RAS/MAPK pathway, and extrinsic (microenvironmental) mechanisms, such as exosome secretion [12, 30, 34-36].
Various phosphatase inhibitors and modulators have been developed to target phosphatase dysregulation in cancer and have shown promising preclinical results (Figure 1) [3]. There are currently 15 phosphatase modulators, mostly targeting SHP2, in the early phases of clinical trials with none being launched [3].

Despite the modulators' therapeutic potential, the design of phosphatase therapeutics is more challenging than the design of TKIs due to the poor understanding of the regulatory system of phosphatases and the structural similarity of phosphatase binding pockets [12, 34, 37]. However, the development of allosteric inhibitors overcame those challenges. For example, SHP099 is an allosteric inhibitor of SHP2 that stabilizes its autoinhibited state, while FIAsh alters SHP2 conformation to prevent substrate binding [38]. In addition to allosteric inhibitors, novel therapeutic strategies for targeting phosphatases are being developed, such as molecules that inhibit the interaction between protein and subunit regulators or endogenous activators [39]. Small molecules and antibodies targeting phosphatases have demonstrated high efficacy in preclinical tumor models and shown synergistic antitumor activity with kinase inhibitors and immune checkpoint blockade [40-46]. However, understanding the roles of phosphatases in both cancer cells and the immune tumor microenvironment is essential for developing effective and selective therapeutics, and tailoring combination therapies for cancer treatment [47].
Phosphatases play diverse roles in cancer, acting as tumor suppressors, oncogenic drivers, or exhibiting dual functionality depending on cellular context [37]. Their dysregulation contributes to tumor progression, therapy resistance, and immune evasion through pathways such as JAK/STAT, MAPK, PI3K, and Hippo signaling pathway [24, 43, 48, 49]. Additionally, phosphatases play a key role in exosome secretion, further modulating the tumor microenvironment (TME) [27-29, 47]. Categorizing phosphatases as tumor promoters, suppressors, or context-dependent regulators provides insight into their complex roles in cancer biology [37]. A deeper understanding of these enzymes will pave the way for novel therapeutic strategies targeting phosphatases in cancer.
This review provides a comprehensive overview of phosphatases in cancer, emphasizing their roles in immune regulation, metastasis, and tumor progression. Additionally, we will explore their involvement in exosome secretion and highlight recent advances in phosphatase-targeting therapies, including preclinical and clinical developments.
2 Classification of Phosphatases in Cancer
Phosphatases are a diverse group of enzymes, with approximately 211 phosphatase domains classified into different families based on catalytic domain similarity [15]. They are categorized into four major classes: phosphoprotein phosphatases (PPP) and phosphatase metal-dependent phosphatases (PPM), which dephosphorylate phosphoserine and phosphothreonine residues; haloacid dehalogenase (HAD) phosphatases, which utilize aspartic acid as a catalytic nucleophile and magnesium as a cofactor; and PTPs, which dephosphorylate phosphotyrosine residues [3, 15].
Humans have seven PPP enzymes, all of which are multimeric: PP1, PP2A, PP2B (calcineurin), PP4, PP5, PP6, and PP7 [50]. For example, PP1 forms a dimeric holoenzyme with catalytic and regulatory subunits, while PP2A consists of a trimeric holoenzyme with catalytic, regulatory, and scaffolding subunits [50]. The multimeric nature of PPP enzymes enables diverse biological functions by regulating posttranslational modifications, subcellular localization, and substrate specificity [3].
PTPs, in contrast, are monomeric enzymes critical for reversing receptor tyrosine kinase (RTK) signaling. They are classified into receptor PTPs (e.g., CD47) and non-receptor PTPs (e.g., PTPN11) [3, 51]. There are about 125 active PTP genes, which encode domains responsible for catalytic activity, subcellular localization, and substrate specificity [52]. PTPs are further divided into cysteine- and histidine-based PTPs, distinguished by the catalytic residue responsible for the nucleophilic attack on substrates [3]. Histidine-based PTPs include two ubiquitin-associated and Src-homology 3 domain-containing (UBASH3) phosphatases, which will not be the focus of this review [53] (Table 1).
| Phosphatase | Phosphatase family | Role in cancer | Pathway affected | Role in cancer |
|---|---|---|---|---|
| PTPN11 (SHP2) | Non-receptor Protein tyrosine phosphatase (PTP) |
|
|
Mostly tumor promoter in cancer cells and immune cells. |
| PTPN6 (SHP1) | Non-receptor protein tyrosine phosphatase (PTP) |
|
|
Tumor suppressor in immune cells |
| PTPN2 (TCPTP) | Non-receptor protein tyrosine phosphatase (PTP) |
|
|
Tumor suppressor in immune cells. |
| Loss is linked to immune evasion and inflammation. | ||||
| PTP1B | Non-receptor protein tyrosine phosphatase (PTP) |
|
|
Tumor promoter in cancer cells. |
| PP2A | Serine/threonine phosphatase (STP) |
|
|
Tumor suppressor in cancer cells. |
| PTPN22 | Non-receptor protein tyrosine phosphatase (PTP) |
|
|
Tumor suppressor in immune cells. |
| JAK/STAT | ||||
| DUSP1/6 | Dual specificity phosphatase (DUSP) | Regulates MAPK signaling by dephosphorylating extracellular signal-regulated kinase (ERK), JNK, and p38. |
|
Dual roles in cancer cell. |
|
||||
| PRL-3 | Dual specificity phosphatase (DUSP) |
|
|
Tumor promoter in cancer cells. |
Among non-receptor PTPs, a notable subfamily is the dual-specificity phosphatases (DUSPs), which dephosphorylate phosphoserine, phosphothreonine, and phosphotyrosine residues [3]. DUSPs are further categorized based on their subcellular localization and substrate specificity. For instance, DUSP1 and DUSP6 are classical DUSPs that regulate the MAPK signaling pathway [54]. Another example is phosphatase of regenerating livers-3 (PRL-3), a pro-metastatic oncogene [55, 56].
While phosphatases such as PTEN and CDC25 have been extensively studied in cancer biology, this review focuses on a subset of cysteine-based non-receptor phosphatases that are emerging as key regulators of cancer progression but remain less explored in the literature [57-59]. Although DUSP3 (VHR) plays a role in MAPK signaling and cancer cell survival, it was excluded from this review due to the limited research specifically addressing its function as a cancer-associated phosphatase [60]. Specifically, we highlight PTPN11, PTPN6, PTPN2, PTP1B, PTPN22, PP2A, DUSP1/6, and PRL3. These phosphatases play pivotal roles in signal transduction, cell cycle regulation, and metastasis, making them promising therapeutic targets in cancer [16-19, 35, 61]. As a result, they are currently being investigated in preclinical and clinical cancer studies [37, 42, 62, 63]. By focusing on these specific phosphatases, we aim to provide a comprehensive look at their emerging roles in cancer, complementing the extensive research on more widely recognized phosphatases.
3 The Role of Phosphatases in Cancer and the Key Signaling Pathways
Phosphatase dysregulation in cancer progression plays two different roles in cancer: tumor suppression or tumor promotion depending on the phosphatases' biological role [3, 64]. Phosphatases such as PTPN6, PTPN2, PP2A, and PTPN22 predominantly function as tumor suppressors by negatively regulating signaling pathways and maintaining cellular homeostasis [3]. These phosphatases negatively regulate the JAK/STAT and TCR immune signaling pathways [3]. Their substrates include Lymphocyte-specific protein tyrosine kinase (LSK) downstream of the TCR, Zeta-chain-associated protein kinase 70 (ZAP-70), and PD-1 [3]. Conversely, phosphatases like PTPN11 (SHP2), PTP1B, PRL3, and DUSP1/6 are primarily associated with tumor-promoting activities, including enhanced proliferative and metastatic signaling [4, 48, 65-67]. PTPN11 positively regulates ERK1/2, and AKT in the MAPK and PI3K oncogenic signaling pathways [68]. Notably, PTPN11 dysregulation and mutations are associated with solid tumors and juvenile myelomonocytic leukemia, respectively [34]. Similarly, PTP1B positively regulates the JAK/STAT and RAS/MAPK pathway [69]. Both PTP1B and PTPN11 dysregulation is associated with poor breast, lung, hepatocellular and gastric cancer [24, 70, 71]. DUSPs are critical for regulating mitogenic signaling pathways such as MAPK and Jun N-terminal kinase (JNK). DUSP1 and DUSP6 substrates include ERK1/2, JNK, and P38 MAPK and its inhibition suppressess leukemia and breast cancer [72-74]. Another phosphatase, PRL-3 promotes tumorigenesis and epithelial to mesenchymal transition by modulating PI3K/AKT activity [41, 75]. PRL3 downregulates the activity of PTEN, a tumor suppressor, thereby activating the PI3K/AKT pathway and promoting metastasis [76]. Furthermore, PRL3 dephosphorylates rhoA and B1 integrin in the src pathway promoting cell migration [35]. PRL3 dysregulation promotes cancer cell survival and modulates the TME through inducing angiogenesis, phenotypic plasticity, and tumor cell crosstalk [35]. PRL-3 prenylation anchors it to the cell membrane, highlighting its key role in the TME and enabling antibody-based targeting [77]. PRL3 dysregulation promotes cancer progression in both tumor and nontumor cells (stromal and cancer-associated fibroblasts) acting as an oncogenic node for various hallmarks of cancer progression [35, 78].
The roles of some phosphatases are highly context-dependent, as some, such as PTPN11 and PP2A, exhibit dual functionality, acting as either tumor suppressors or promoters depending on the cancer type, genetic background, and microenvironmental factors [18, 25]. This duality highlights the complexity of phosphatase biology and underscores the need for context-specific therapeutic approaches [3, 17, 35]. For example, SHP2 has been reported to have tumor suppressive function by acting on the JAK/STAT3 pathway reducing inflammation [25, 79-81]. PP2A suppresses tumor growth by regulating the MAPK and PI3K/AKT signaling pathways. In contrast to the tumor-suppressive roles of the PP2A, the PP2A subunits striatin-3 (STRN3) and striatin-4 (STRN4) exhibit oncogenic roles [82]. It inhibits key oncogenic effectors, including ERK1/2, AKT, and c-Myc, thereby suppressing tumor progression [26, 83]. Studies reported that inhibitors and activators of PP2A suppress tumorigenesis, suggesting the important role PP2A plays in promoting tumor cell survival [22, 23, 84]. Overall, phosphatases exhibit complex and dynamic interactions with signaling pathways. Understanding their molecular mechanisms in the cellular contexts will provide insight into their roles in cancer, where they can function as tumor suppressors, oncogenic drivers, or context-dependent regulators.
4 Phosphatase Dysregulation in Cancer: Intracellular Effects
Phosphatases play a critical role in cancer growth, acting as tumor suppressors or tumor promoters or have dual functions. In cancer cells, they modulate oncogenic signaling pathways such as MAPK, PI3K, and Hippo in cancer cells [85-87], while in immune cells, they modulate the JAK/STAT and PD1 signaling pathways [31, 88]. The role of phosphatases in cancer depends on their localization and the specific cell type in which they are expressed, further complicating their functions in tumor development and progression [37]. For example, SHP1 is expressed in immune cells and is generally known as a tumor suppressor [19]. The expression of negative SHP1 led to an increase in myeloid colony formation in embryonic stem cells [89]. Furthermore, SHP1 deficient mice developed enlarged neutrophils and monocyte cells and increased macrophage proliferation in peripheral blood [90]. In line with these findings, the epigenetic loss of function mutation of SHP1 is associated with various malignancies such as lymphoma and leukemia, and thus SHP1 known to be a tumor suppressor [91]. Furthermore, the dysregulation of PTPN22 promotes immunosuppression due to its expression in immune cells [92]. While, SHP2 drives both cancer cell proliferation and immunosuppression as it is expressed in both tumor and immune cells [24, 93]. Furthermore, tumor promoters such as SHP2, PTP1B, and DUSP1/6 promote proliferation and survival of cancer cells via the activation of the MAPK signaling pathway [18, 94, 95]. Phosphatases with dual roles in cancer cells, such as SHP2 and PP2A, modulate the MAPK and PI3K/AKT signaling pathways, thereby promoting tumor growth [22, 96, 97]. On the contrary, SHP2 and PP2A exhibits tumor-suppressive functions via the STAT3 pathway in cancer cells, and through forming complexes with endogenous inhibitors, particularly the B55 and B56 subunits, respectively [22, 98]. PRL3 plays a critical role in metastasis and cancer plasticity by localizing to the cell surface, where it interacts with molecules like RhoA, highlighting the impact of cellular localization on phosphatase function [35, 65].
4.1 SHP1 in Tumor Suppression and Therapeutics
SHP1 contains a PTP catalytic domain and two SH2 regulatory domains (N-SH2 and C-SH2), along with a C-terminal tail [63]. SHP1 and SHP2 are autoinhibited by their N-SH2 domain, blocking their catalytic activity [9]. SHP1's dysregulation is associated with several malignancies such as lymphomas and leukemias. They are expressed in both hematopoietic and epithelial cells, with SHP1 localized in the cytosol and nucleus of hematopoietic and epithelial cells, respectively [3, 99]. SHP1 has a well-known negative regulatory function in immune cells, specifically targeting T cells, including TCR, zeta chain-associated protein of 70 kDa (ZAP70), and phosphoinositide 3-kinases (PI3K) reducing T-cell activity [99, 100]. Activation of SHP1 by TNF-α has been reported to reduce endothelial cell proliferation in response to growth factors such as VEGF by dephosphorylating ERK and the MAPK pathway [101]. This suggests that SHP1 acts a tumor suppressor in both endothelial cells and immune cells. Furthermore, in hepatic stellate cells, SHP1 negatively regulates the proliferative response by reducing the expression of cyclin D1 through the dephosphorylation of AKT and ERK1/2 kinases [102]. Similarly, SHP1 downregulates oncogenic STAT3 pathway hyperactivation, which is involved in signaling for proliferation, cell survival, invasion, and angiogenesis (VEGF) [103-105].
In immune cells, SHP1 negatively regulates cytokine receptors by dephosphorylating substrates in the JAK/STAT pathway [106]. SHP1 downstream of granulocyte macrophage colony-stimulating factor receptor (GM-CSFR) inactivates RAS, STAT3, and MAPK inhibiting overall myeloid proliferation and survival [100]. SHP1 acts downstream of the immunoreceptor tyrosine-based inhibition motif (ITIM) and immunoreceptor tyrosine-based switch motif (ITSM) domains on PD-1 in T cells, activating the PD-1 checkpoint receptor [100]. Furthermore, SHP1 is recruited by signal regulatory protein (SIRPa) suppressing phagocytosis in tumor associated macrophages [107]. SHP1 mutants exhibit hyperactivation of macrophages, dendritic cells, and neutrophils [19, 108]. Tumor-associated mutations in SHP1, including epigenetic silencing, have been identified in various cancers such as uterine carcinoma, ovarian cancer, hepatocellular carcinoma (HCC), and melanoma [91]. SHP1 expression is inversely proportional in patients with AML and glioblastoma [109]. Similarly, SHP1 overexpression is correlated with high survival rates in patients with breast, esophageal, hepatocellular, and prostate cancers [110]. SHP1 dysregulation in leukemias and lymphomas occurs through promoter methylation, alternative splicing, ubiquitination, and loss-of-function mutations [90, 111, 112]. These findings suggest that SHP1 regulates the proliferation of hematopoietic cells, acting as a tumor suppressor, and its deficiency is associated with hematological malignancies. In contrast, a few studies have reported that SHP1 expression is inversely correlated with survival in patients with acute myeloid leukemia (AML), colorectal cancer, and glioblastoma [110]. Thus, SHP1 is generally regarded as a tumor suppressor, but its role may depend on the tumor type. However, the mechanism of its tumor-promoting function has yet to be elucidated.
SHP1 inhibition has been reported to be promising for cancer therapy, as it negatively regulates the immune checkpoint blockade and cancer cells [103]. A study reported that sorafenib and regorafenib activated SHP1 and suppressed TGF-β1-induced epithelial to mesenchymal transition in HCC and lung cancer, respectively [3, 113]. Studies have demonstrated that SHP1 inhibition is beneficial for adoptive T-cell immunotherapy because SHP1 is necessary for the tumor cell non-lysis phenotype [114]. Another In Vitro study demonstrated that SHP1 knockdown enhanced antitumor activity of T cells, an effect that was exacerbated by PD-1 and CTLA-4 blockade [5, 105]. Additionally, SHP1 depletion reduced the immunosuppression of regulatory T (Treg) cells in the tumor microenvironment [114]. However, the reduction of SHP1 in TILs restored their function In Vitro [115]. These studies demonstrate that targeting SHP1 in immunotherapy induces a pro-inflammatory response across various immune cell populations [5, 114, 115]. Although targeting SHP1 is a promising immunotherapeutic strategy, it may lead to severe side effects. However, it could offer benefits for adoptive cell transfer therapy by inducing a robust antitumor response while minimizing systemic toxicities [5].
4.2 PTPN22 in Tumorigenesis and Therapeutics
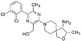
PTPN22 is a PTP expressed in hematopoietic cells and is characterized by an N-terminal PTP domain, interdomain, and C-terminal domain that binds to CSK [3]. While the catalytic domain of PTPN22 is well defined, the structural details of the other regions remain unclear, complicating the specific targeting of PTPN22 for therapeutic purposes [116]. PTPN22 plays a negative regulatory role in immune cells by inhibiting T-cell receptor (TCR) signaling through dephosphorylation of ZAP70 and the TCR. Additionally, it inhibits the immunosuppressive functions of Treg cells and affects B cells, dendritic cell signaling, and inflammasome activation by dephosphorylating NLR family pyrin domain 3 (NLRP3) [3, 61]. PTPN22 deficiency in T cells enhances the efficacy of TGF-β antibodies [117]. Deletion of PTPN22 also enhances the effectiveness of anti-PD-L1 therapy in tumor models, as evidenced by the high CD8+ T-cell to Treg cell ratio. Furthermore, the absence of PTPN22 without immune checkpoint therapy led to elevated levels of CD4+ and CD8+ T cells, macrophages, and NK cell infiltration In Vivo, and suppressed overall tumor growth [92]. Another study demonstrated that PTPN22-deficient T cells improved lymphoma tumor clearance in mice [118]. Additionally, PTPN22-deficient CD8+ T cells produced high levels of granzyme B and IFN-γ when cocultured with ovarian carcinoma cells In Vitro [117]. Moreover, PTPN22 deficiency was associated with enhanced memory T cell function, characterized by increased secretion of IFN-γ and TNF, which improved their tumor-killing capabilities and persisted after initial tumor clearance [119]. These findings underscore the importance of targeting PTPN22 to improve antitumor immunity.
A 6-hydroxybenzofuran-5-carboxylic acid scaffold (IC-11) has been identified as a PTPN22 inhibitor with an IC50 of 4.6 ± 0.4 µM [120]. IC-11 increased TCR-stimulated Lck394 and ERK1/2 phosphorylation in cells expressing wild-type PTPN22, suggesting that IC-11 modulates T-cell signaling through PTPN22 inhibition [120]. Other derivatives of IC-11, such as A-15 and A-19, displayed high specificity and have been shown to modulate the TCR-mediated expression of IL-2. NC-1, a noncompetitive inhibitor of PTPN22, increased p-ERK activation with no effect on T cells lacking PTPN22 expression, indicating TCR-mediated activation through PTPN22 inhibition [61]. Another inhibitor, LTV-1, enhanced TCR signaling in a dose-dependent manner and increased Lck-394 and ζ chain phosphorylation in Jurkat cells [121]. L-1, a catalytic small-molecule inhibitor, exhibited higher specificity for PTPN22 compared to other PTPs [3, 61, 122]. Notably, L-1 is the only PTPN22 inhibitor reported to exhibit pharmacokinetic activity In Vivo. The catalytic inhibitor L-1 significantly reduced tumor growth in tumor models, and this effect was more pronounced in combination with anti-PD-L1 therapy. PTPN22 inhibition in combination with anti-PD-L1 increased macrophage and CD8+ and CD4+ T-cell tumor infiltration [61]. The antitumor activity of L-1 is mediated by tumor-associated macrophages, as the depletion of macrophages reduced the efficacy of L-1. A recent study demonstrated pTYR mimetics (D34 and D14) to reversibly inhibit PTPN22 activity Ki = 0.93 and 1.34 μM, respectively. They specifically inhibit TCR signaling which reduced macrophage (M2) polarization and enhanced T-cell activation. Furthermore, D34 and D14 had a synergistic antitumor effect with PD1 blockade [123]. These findings suggest that PTPN22 is a promising immunotherapeutic target for cancer treatment, both as a monotherapy and in combination with immune checkpoint inhibitors [51, 59].
4.3 DUSP1 and DUSP6 in Tumorigenesis and Therapeutics
DUSP1 and DUSP6 are DUSPs that inactivate the MAPK signaling pathway through the dephosphorylation of threonine and serine residues [124]. DUSP1/6 is crucial for regulating the MAPK, MEK, and JNK pathways, and their deletion results in uncontrolled activation of these oncogenic signaling pathways [125]. Both DUSP6 and DUSP1 interact with a MAPK-binding domain and kinase-interacting segment. DUSP6 specifically interacts with ETS-1 and c-JUN transcription factors, which are involved in basal-like breast cancer [124]. DUSP6 primarily dephosphorylates ERK1/2 [125], while DUSP1 can dephosphorylate ERK1/2, p38, and JNK, promoting survival signaling (Figure 2A) [95, 126]. DUSP1 has been reported to promote chemo- and radio-resistance in breast cancer cells and reduce UV-induced apoptosis in leukemia cells by modulating the JNK pathway [6, 127]. High DUSP1 and DUSP6 expression levels are associated with poor prognosis and overall survival in non-small cell lung cancer (NSCLC) [128, 129]. Similarly, DUSP6 has been linked to uncontrolled proliferation, metastasis, and poor prognosis in gastric cancer [95]. Another study demonstrated that DUSP6 inhibition reduced metastatic dissemination in NSCLC In Vivo [130]. Similarly, another study demonstrated that DUSP6 inhibition stopped the transformation of B cells to B-acute lymphoblastic leukemia (B-ALL) via the ERK pathway [95]. Additionally, DUSP6 inhibition exhibited synergistic effects with cisplatin in patient-derived xenograft (PDX) mouse models [126]. Moreover, DUSP inhibition enhanced the activity of the JAK/STAT inhibitor ruxolitinib and suppressed colony formation in B-ALL cells and increased the survival rates of In Vivo models [6, 127]. The best-characterized DUSP1/6 allosteric inhibitor is BCI, which acts as a dual inhibitor by binding to an allosteric pocket in DUSP and induces a conformational change that prevents it from binding to its substrates (Figure 2B) [131]. BCI selectively inhibits DUSP1 and DUSP6 without affecting other phosphatases such as Cdc25B, PTP1B, or DUSP3 In Vitro [127]. Additionally, BCI increased tumor cell apoptosis in a patient-derived xenograft B-ALL mouse model [132]. Similarly, another In Vitro study demonstrated that DUSP1 inhibition abolished imatinib resistance in a BCR-ABL PDX model [74]. These findings suggest that DUSP1 and DUSP6 inhibitors are promising targets for cancer therapeutics, with potential applications in combination therapies to enhance antitumor immunity.

4.4 PRL3 in Tumorigenesis and Therapeutics
PRL-3 is a novel class of PTPs with a C-terminal prenylation motif, and it is primarily found in cardiac and skeletal muscle [77]. PRL-3 is a dual-specific phosphatase implicated in various malignancies, including breast, colon, and prostate cancers [75]. PRL3 overexpression is also associated with hyperactivity of the EGFR signaling pathway in various cancer cell lines (Figure 3A) [133]. PRL3 is hypothesized to activate the Src pathway and has been found to directly interact with MYC in T-cell acute lymphoblastic leukemia (T-ALL), where its inhibition suppressed tumor growth In Vivo [65, 75]. PRL3 enhances APC/FZR1 activity by dephosphorylating FZR1, driving colorectal cancer progression [134]. Stable overexpression of PRL3 has been associated with increased cell migration and invasion in CHO cells, as well as cancer metastasis In Vivo, with similar findings observed in liver, gastric, and prostate malignancies [130, 135-138]. Clinically, PRL3 overexpression has been reported in various metastatic cancers, including prostate, esophageal, and NSCLC [75]. Furthermore, PRL3 promotes tumorigenesis through its interaction with membrane-associated magnesium ion channels, CNNM3/4 [139, 140]. The mutation of CNNM-binding segment prevented PRL3 binding, which abolished metastasis in B16 melanoma cells [140]. In addition to modulating metastasis, PRL3's interaction with CNNM also modulates metabolic plasticity, allowing it to meet the metabolic demands needed for high proliferation rates [141].

A study reported PRL3 induces a mesenchymal phenotype in cancer cells, which suggests PRL3 has a role in the early stages of metastasis [141]. Furthermore, PRL3 expressed on tumor cell surfaces enhanced cancer cell adhesion to extracellular matrix substrates such as fibronectin and laminin [136, 141]. A study reported that coculturing PRL3+ colorectal cancer (CRC) cells with tumor-associated macrophages (TAMs) promoted tumor cell invasion, implicating crosstalk between PRL3-expressing cancer cells and macrophages [142, 143]. The PRL3 inhibitor JMS-053 is a reversible, noncompetitive drug with an IC50 value of 18 nM (Figure 3B). It binds to an allosteric pocket in PRL3, preventing conformational change, but it lacks specificity, as it inhibits both PRL2 and PRL3 [144]. To address this, a derivative called NRT-970-59 was developed with high specificity and potent inhibitory effects in various cancer lines [145]. In ovarian cancer, NRT-970-59 reduced migration, disrupted spheroid growth, and decreased RhoA activity in ovarian cancer cells, suggesting that targeting PRL3 has significant antitumor efficacy [146]. Furthermore, the expression of PRL3 on tumor cell surfaces allows it to be targeted using monoclonal antibodies, which has the advantage of higher specificity compared to small-molecule inhibitors [145]. Similar to SHP2, PRL3 plays a vital role in cancer plasticity, making it a promising therapeutic target.
4.5 The Dual Role of PTP1B in Cancer and Therapeutics
PTP1B has three domains: N-terminal catalytic domain, a regulatory domain, and a C-terminal domain, which functions to localize the phosphatase to the endoplasmic reticulum membrane [147]. PTP1B PTP regulates growth factor and hormone signaling and is associated with the progression of various cancers, including breast, prostate, gastric, and colorectal cancers [148]. It dephosphorylates Src at Y530 and p62Dok, thereby activating the RAS/MAPK and PI3K/AKT pathway [13]. Oncogenic PTP1B is overexpressed in tumors, and this overexpression is linked to its interaction with other signaling pathways [149]. For example, PTP1B overexpression is associated with poor prognosis in pancreatic ductal adenocarcinoma (PDAC) patients [150]. Furthermore, its overexpression in PDAC cell lines promoted high proliferation rates and migration of the tumor cells. These results were supported by In Vivo studies demonstrating that PTP1B knockdown or inhibition significantly reduced tumor growth. PTP1B promotes tumorigenesis via the pyruvate kinase M2 (PKM2) pathway, which is involved with glucose metabolism, proliferation, and angiogenesis [150, 151].
Additionally, PTP1B activates the JAK/STAT pathway by dephosphorylating JAK1/3, STAT1, STAT3, and STAT5 [152]. PTP1B deficiency in T cells has been reported to inhibit tumor growth in various malignancies, including mammary, melanoma, and colorectal tumors [148]. These findings demonstrated that targeting PTP1B enhances T-cell antitumor immunity creating an overall pro-inflammatory tumor immune microenvironment (TIME). Studies have shown that PTP1B deletion in T cells enhances CD8+ T cell expansion and cytotoxicity in the TIME of solid tumors by amplifying STAT5 signaling [148]. Furthermore, PTP1B degrades the stimulator of interferon genes (STING) via the proteasome, thereby abolishing its inflammatory response [153, 154]. Another In Vitro study demonstrated that PTP1B deletion enhanced T-cell activity by increasing their sensitivity to IFN cytokines [155]. Thus, PTP1B modulates immune signaling pathways distinct from those mediated by SHP1, such as the PD-1 and CTLA-4 pathways, enabling the targeting of multiple immune cell types [101, 148].
PTP1B also has a tumor suppressor role through dephosphorylating oncogenic molecules such as src, Ras, and PI3K that are induced by growth factors. PTP1B overexpression was shown to reduce tumorigenecity in v-src transformed cells [156]. Another study demonstrated that PTP1B upregulation abolishes BCR-ABL induced transformation and the differentiation of BCR-ABL-expressing cells [157]. Furthermore, PTP1B suppressed the oncogenic effects of v-src, v-crk, and v-ras. These studies suggest that PTP1B inhibits tumorigenesis by acting upstream of Ras and suppressing ERK activation [158]. In addition, PTP1B has been reported to promote apoptosis by reducing RTK signaling, enhancing ER stress signaling, or activating caspase-8 and -9 [158]. Another study demonstrated that PTP1B reduced apoptosis through upregulating AKT phosphorylation and lowering FOXO1 expression [159]. PTP1B induces apoptosis through multiple mechanisms, including promoting the unfolded protein response downstream of ER stress via the IRE1 pathway and promoting caspase-mediated cell death. A study demonstrated that PTP1B activates caspase-8 and caspase-9, leading to cell death in glioma cells following treatment with the PPAR-γ agonist troglitazone [160]. Another proposed mechanism by which PTP1B induces apoptosis is through the suppression of pro-survival STAT3 signaling, thereby increasing cell sensitivity to caspase-driven cytotoxicity following drug treatment [161]. Another tumor suppressor role of PTP1B is reducing metastasis by regulating cadherin-mediated cell contact [162]. Notably, wild-type PTP1B associates with N-cadherin and dephosphorylates β-catenin, while catalytically inactive PTP1B reduces cell-to-cell adhesion, thereby promoting cell migration and invasion [162]. Another study demonstrated PTP1B deletion in p53-mutant mice increased tumorigenesis and reduced survival rates [163].
Although PTP1B has a dual function in tumors, the therapeutic strategy for targeting PTP1B in cancer is through inhibition. A dual inhibitor for PTP1B and PTPN2 (1B/TC) improved monocyte dendritic cell maturation (moDC) and when injected into mouse models it suppressed tumor growth, and enhanced IFN-γ secretion from T cell co-cultures derived from pancreatic cancer patients. An analog of MSI-1436, DPM-1001, was developed with high potency, and its activity is enhanced by binding to copper; however, its mode of action requires further elucidation. Similarly, another small-molecule inhibitor, compound 182, enhanced T-cell recruitment and activation, suppressing tumor growth In Vivo with minimal toxicity [164]. Overall, these findings indicate that PTP1B is a promising immunotherapeutic target for cancer treatment, both as a monotherapy and in combination with immune checkpoint inhibitors. Modulation of PTP1B activity enhances antitumor immunity and has the potential to improve prognostic outcomes in various cancer types. However, understanding the dual role of PTP1B is crucial for appropriately targeting it in tumors.
4.6 The Dual Role of PTPN2 in Cancer and Therapeutics
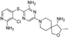
PTPN2 is a ubiquitous PTP that exists in alternative splice variants, notably TC-45 and TC-48 [3]. TC-45 translocates into the nucleus and, in response to extracellular signaling such as epidermal growth factor (EGF), TNF, and IFN-γ, moves into the cytoplasm where it interacts with various substrates at the plasma membrane, such as EGFR, JAK1/2, and STAT1/3 [165]. TC-48 has a hydrophobic C-terminus that localizes to the endoplasmic reticulum, allowing it to dephosphorylate-specific substrates. PTPN2 dephosphorylates KRAS, which affects its localization to the plasma membrane and activates the MAPK downstream signaling [21]. PTPN2 knockdown significantly reduced the proliferation in KRAS-dependent cancer cells but not in KRAS wild-type cancer cells, indicating the dependence of KRAS mutations on PTPN2 to modulate its downstream signaling pathway for cancer survival [21]. PTPN2 also promotes mitochondrial oxidation human colorectal cancer cells through the phosphorylation of STAT3 [166]. Whereas its deletion decreased ATP production and increased cancer cell survival and migration [166]. In addition, PTPN2 expression is associated with cancer incidence, as high levels of PTPN2 have been observed in gastric cancer cell lines and overexpressed in CRC tumor tissues [167]. Similarly, high PTPN2 transcript levels are found in isocitrate dehydrogenase (IDH) wild type and mesenchymal subtypes of gliomas [168]. PTPN2 promotes tumorigenesis in glioma through promoting oxidative stress and inflammation cytokines [169]. On the contrary, PTPN2 loss enhanced SFK and STAT3 signaling, promoting tumorigenicity in breast cancer In Vitro and In Vivo [170].
PTPN2 plays a significant role in the TIME by inhibiting responses to pro-inflammatory cytokines, such as IL-2, IL-6, IL-15, and IFN-γ, through the dephosphorylation of components in the JAK/STAT signaling pathway [3]. Furthermore, PTPN2 inhibits TCR signaling by dephosphorylating the activation motifs of Src family kinases [171]. Deletion of PTPN2 in T cells enhanced the cytotoxicity of CD4+ Th1 cells and CD8+ T cells through the activation of JAK/STAT signaling and increased IFN-γ secretion [3, 172, 173]. Similar reports demonstrated that PTPN2-null T cells protected TP53 heterozygous mice from tumor growth and abolished breast cancer implants by activating CD4+ and CD8+ effector and memory cells [3, 173]. High PTPN2 levels were found to significantly increase immune cell proliferation, such as macrophages, neutrophils, and CD8+ cells [168]. Moreover, PTPN22 inhibition primed antitumor immunity in tumors by sensitizing cancer cells to IFN-γ [173]. ABBV-CLS-484 (AC484) is a PTPN2/N1 catalytic inhibitor that exhibits potent antitumor activity In Vitro. AC484 enhanced JAK/STAT signaling activity, promoting CD8+ and natural killer cells activity, thereby improving overall T-cell function and fostering an inflammatory tumor microenvironment [173].
4.7 The Dual Role of PP2A in Cancer and Therapeutics
PP2A is a heterotrimeric phosphatase composed of three distinct subunits: the catalytic subunit C (Cα), the scaffold subunit A (PR65α), and the regulatory subunit B (B55/56). This heterotrimeric phosphatase exhibits both oncogenic and tumor-suppressor functions, depending on the complex it forms [82]. The regulatory subunit encompasses multiple subfamilies, such as B55 and B56, which regulates PP2A activity and its cellular localization. The B55 and B56 subunits act as tumor suppressors, counteracting the endogenous proteins (CIP2A and SET) that drive tumor progression (Figure 4A). The overexpression of CIP2A and SET promotes tumor progression by inhibiting PP2A activity, stabilizing c-Myc, and activating the PI3K pathway [174]. In contrast to the tumor-suppressive roles of the PP2A subunits B55 and B56, the PP2A subunits striatin-3 (STRN3) and striatin-4 (STRN4) exhibit oncogenic roles. They act as scaffolds between PP2A and kinases, inhibiting PP2A activity in tumor-suppressive pathways, and redirect its activity toward oncogenic pathways instead [82].

PP2A functions as a tumor suppressor primarily by inhibiting key oncogenes, including MYC, ERK, AKT, and BCL-2 in the MAPK and PI3K/AKT pathways [22, 175]. Dysregulation of PP2A's inhibitory function has been reported in various malignancies due to PP2A mutations, dysregulation of its C-terminal tail phosphorylation, or the overexpression of its regulators, such as CIP2A and SET [176]. PP2A/SET complex is overexpressed in chronic lymphocytic leukemia (CLL) and AML [177]. In addition, PP2A inactivation is reported in breast cancer and is associated with poor prognosis [178].
PP2A also plays a role in promoting metastases of tumor cells through modulating YAP and TAZ, oncogenic transcription factors, activity. YAP and TAZ are tightly regulated by kinases such as MST1/2, while the PP2A-SRN3/SRN4 complex dephosphorylates the tumor-suppressive Hippo kinases MST1/2 and MAPK4, leading to the activation of the oncogenic YAP protein [44, 179].
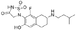
CC11 and CM-1231 are small molecules that target PP2A activity by inducing a conformational shift in the SET protein, thereby enhancing its antitumor activity [177, 180] (Figure 4B).
A study demonstrated that CC11 and CM-1231 inhibited leukemia tumor growth by promoting apoptosis in CLL and AML cells, respectively [177, 180]. Furthermore, CC11 induced mitochondrial apoptosis through PP2A-mediated direct dephosphorylation of the proapoptotic BAD protein. Similarly, CM-1231 inhibited the interaction between PP2A and SET, inducing apoptosis in AML cell lines and primary patient samples [180]. Other small-molecule activators of PP2A (SMAPs), which are derivatives of phenothiazine, have also been reported to stabilize PP2A function. Perphenazine suppressed T-ALL growth and induced apoptosis by dephosphorylating BAD and AKT in primary human T-ALL cells In Vivo. These findings demonstrate that reactivating PP2A has strong antitumor activity In Vivo but exhibited toxicity at the inhibitory dose level [181, 182]. SMAPs work by enhancing B55 and B56 subunit affinity to the PP2A dimer [22]. However, more research is needed to elucidate the precise mechanism of action of SMAPs, as they have been reported to bind to the PP2A dimer without the B-subunit, suggesting an alternative mode of action [22].
PP2A reactivation overcame kinase therapy resistance, sensitizing cancer-resistant cells to imatinib in myeloid leukemia cells [183]. Fingolimod is an FDA-approved drug that was reported to target PP2A and has demonstrated potent antitumor activity. Fingolimod (FTY720) triggers a conformational change that releases SET from PP2A-C, reactivating the phosphatase [184]. FTY720 treatment has shown beneficial effects in hematological malignancies by inhibiting BCR/ABL activity modulated by PP2A in myeloid and lymphoid cell lines [183, 185]. Notably, FTY720 decreased cell growth, promoted apoptosis, and lowered ERK and AKT activity in breast cancer in both In Vitro and In Vivo models. However, CM-1231 was more potent than FTY720 in reducing metastases with minimal cardiotoxic effects, while FTY720 induced cardiotoxicity in AML zebrafish models [180].
Although the primary focus of PP2A therapeutics is to activate its tumor suppressor activity, inhibiting the catalytic subunit of PP2A was proven to be effective for cancer cell killing, particularly for enhancing antitumor immunity [186].
PP2A primarily functions as a tumor suppressor in cancer cells, but it is also an emerging target for immunotherapy due to its role in suppressing T cell activity. A study reported that silencing PP2A gene led to T-cell accumulation in B16-ova tumors, inhibited apoptosis, increased proliferation rates, and enhanced the production of pro-inflammatory cytokines, such as interferon (IFN) and IL-2 [187, 188]. Similarly, other studies have demonstrated that silencing PP2A in CD4+ or CD8+ T cells enhanced their antitumor activity in In Vivo mouse models, with PP2A knockdown increasing IFN-γ secretion from CD8+ T cells [187].
A study reported that PP2A inhibitor LB-100 enhanced immune checkpoint blockade activity in In Vivo mouse models [189]. Specifically, LB-100 in combination with anti-PD-1 therapy suppressed tumor growth and prolonged survival rates in colorectal cancer mouse models compared with single-agent treatments [190, 191]. LB-100 in combination with anti-PD-1 increased T cell infiltration and activation and demonstrated high efficacy in models of B16 melanoma and glioblastoma. In coculture experiments, LB-100 enhanced IFN-γ secretion from CD8+ T cells, which increased expression of PD-L1 in glioblastoma cells, suggesting PP2A in combination with anti-PD1 is more efficacious than single-agent therapy [191]. Furthermore, PP2A promoted Treg suppressive functions in the TIME by increasing IL-2 receptor expression and inhibiting the mTOR signaling pathway [192]. Overall, PP2A has been studied for cancer cell sensitization to chemotherapy and immunotherapy [22, 23, 84, 185, 193]. Its multimeric structure presents challenges for targeting. However, by targeting specific heterotrimers or inhibiting its interactions with regulators, it is possible to effectively modulate PP2A activity [45]. Further research is needed to elucidate the role of PP2A heterotrimers in cancer and the immune tumor microenvironment.
4.8 The Dual Role of SHP2 in Cancer and Therapeutics
SHP2 is a PTP that is widely present in all tissues and functions downstream of various membrane receptor tyrosine kinases (RTKs) and plays a critical role in immunosuppression [194]. Similar to its homolog SHP1, SHP2 possesses tandem SH2 domains (N-SH2 and C-SH2) and a PTP catalytic domain, along with a disordered C-terminal tail [195]. Similar to PTP1B, SHP2 modulates the activity of several receptor tyrosine kinases, including EGFR and MET, as well as the JAK/STAT pathway, thereby activating their downstream signaling pathways [40]. SHP2 activates GRB2 and acts as a scaffold for GAB2 and tyrosine kinase receptors, promoting myeloid transformation via RAS–Raf–MEK signaling pathway activation, which leads to aberrant proliferation, migration, and survival in AML cells (Figure 6A) [47, 196]. SHP2 mediates tumor progression by activating PI3K/AKT pathway, as evidenced by a study reporting that SHP2 E76K mutant significantly promoted chemoresistance in bone marrow stromal cells and upregulated vascular cell adhesion molecule 1 (V-CAM-1) through PI3K/AKT signaling in B-precursor acute lymphoblastic leukemia [197]. Furthermore, SHP2 plays a crucial role in the Hippo signaling pathway by modulating YAP1, which has been associated with tumor progression [198]. Additionally, the nuclear translocation of YAP1 is facilitated by SHP2's dephosphorylation of Tyr357 in YAP1, triggering the expression of metastatic genes [198, 199]. Notably, the nuclear co-localization of YAP1 and SHP2 correlates with poor prognosis in NSCLC, underscoring the significant roles of SHP2 and YAP in metastasis [198]. Furthermore, SHP2 dephosphorylates parafibromin in the nucleus, leading to the activation of TCF/LEF and TEAD target genes. These genes interact with YAP1 to promote the expression of proliferation- and metastasis-related genes, therby promoting cancer cell growth [200]. These findings suggest that SHP2 inhibition could be used in combination with TEAD inhibitors, presenting a novel therapeutic strategy.
SHP2 induces immunosuppression by negatively regulating pro-inflammatory transcription factor expression and overall cytotoxic T cell activity and M2 macrophage differentiation [201-203] (Figure 5). SHP2 promotes myeloid cell differentiation into an anti-inflammatory phenotype, suppresses cytotoxic T cell differentiation, and reduces pro-inflammatory cytokine expression [201, 204]. Similar to SHP1, SHP2 also promotes immunosuppression in the TIME by positively regulating immune checkpoint proteins such as PD-1, PD-L1, and PD-L2 [205]. The ITSM and ITIM elements are recognized by the C-SH2 and N-SH2 domains of SHP2, respectively, leading to the dephosphorylation of the TCR, which reduces T cell activation and promotes immunosuppression [201, 204]. PD-1 activation is primarily mediated by the phosphorylated SHP2-PD-1 complex, which dephosphorylates ZAP70, PKCθ, and PI3K substrates downstream of TCR. SHP2 dephosphorylation of those substrates disrupts the interaction of immunoreceptor tyrosine activation motifs (ITAM) with TCR, inhibiting the MAPK and PI3K pathways and suppresses overall T-cell activity [201]. Furthermore, PI3K activation and AKT phosphorylation through the PD-1–SHP2 complex suppresses T-cell nuclear factor, activator protein, and pro-inflammatory transcription factor synthesis, inhibiting overall T-cell activity [206]. SHP2 regulates the JAK/STAT pathway by intracellularly dephosphorylating STAT downstream of the immune receptor [207]. Depending on the specific STAT protein it dephosphorylates, SHP2 can either activate or repress the JAK–STAT pathway. For example, SHP2 downregulates STAT1 phosphorylation, reducing MHC II transcription, which is essential for lymphocyte differentiation [207]. The downregulation of p-STAT1 mediated by SHP2 overexpression reduces antigen recognition in CD4+ cells, leading to immunosuppression in the TIME [207]. Conversely, a study demonstrated that SHP2 ablation increases liver cancer development through STAT3 removal in HCC cell lines. Similarly, another study reported a tumor suppressor role for SHP2 in glioma and esophageal tumor via the STAT3 pathway. SHP2 degradation of ubiquitin-conjugated SHP2 promoted glioma proliferation [98]. Additionally, SHP2 overexpression decreased cellular proliferation, glycolysis, and STAT3 phosphorylation in glioblastoma multiforme (GBM) [98] These findings suggest that SHP2 plays a dual role in cancer that is context dependent, it acts as a tumor suppressor when it downregulates the STAT3 pathway. But is a tumor promoter when it activates other pathways such as MAPK and PI3K/AKT.

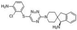
Currently, 15 SHP2 allosteric inhibitors are being tested in Phases 1 and 2 clinical trials, both as monotherapies and in combination with other therapeutics [206]. The scaffold design of these allosteric inhibitors is a critical factor in the clinical trial outcomes of SHP2 therapeutics in cancer patients [95]. Three allosteric sites have been identified in SHP2: the tunnel, latch, and groove, with the tunnel pocket being the most targeted by allosteric inhibitors [38]. SHP099 was the first allosteric inhibitor, paving the way for other inhibitors, including RMC-4640, TN0155, and JAB-3068, and are currently being investigated in clinical trials (Figure 6B) [208].

RMC-4550 is approximately 100-fold more potent than SHP099 and exhibits high bioavailability and efficacy in several malignancies, including tumors bearing KRAS G12C, NF1 loss-of-function, and BRAF Class 3 mutations [203]. It functions as a “molecular glue,” stabilizing the autoinhibited conformation of SHP2 by locking the N-SH2 domain to the catalytic domain [94, 98, 99]. A study showed that SHP099, an allosteric inhibitor, depletes M2 macrophages and induces tumor secretion of CCL5 and CXCL10 chemokines, which recruits T cells and increases T- and B-lymphocyte levels in KRAS and EGFR NSCLC models [209]. However, SHP099 treatment alone increased the number of immunosuppressive granulocytic myeloid-derived suppressor cells (gMDSCs). When combined with a CXCR1/2 inhibitor, SHP099 ameliorated gMDSC infiltration, yielded high CD8+ effector cells with enhanced cytotoxicity, and improved overall survival in KRAS and EGFR mutant models, suggesting that SHP2 combination therapy is more beneficial than monotherapy [209]. Furthermore, RMC-4550 induced antitumor immunity equivalent to or greater than that induced by immune checkpoint blockade. RMC-4550 reduces immunosuppressive M2 macrophages and increases pro-inflammatory M1 macrophages through a mechanism independent of IFN-γ and CD8+ T cells [210]. JAB-3068, an SHP2 allosteric inhibitor, inhibited the PD-1 signaling pathway, attenuated immunosuppression, and induced antitumor activity [40]. JAB-3068 exhibited strong antitumor activity in NSCLC, colorectal cancer, and esophageal cancer cell lines when combined with an anti-PD-1 antibody.
A new type of SHP2 allosteric inhibitor (FIAsh) has been reported that binds to a cryptic allosteric pocket in the catalytic domain, inducing a conformational change that renders the catalytic site inaccessible to substrates. FIAsh has been shown to inhibit SHP2 activity in a dose-dependent manner and displays high selectivity over other phosphatases [202]. SHP2 therapeutics have various allosteric inhibitors that differ in their scaffold design, but target the same tunnel allosteric pocket [47]. SHP2 allosteric inhibitors such as SHP099 have reported scaffold-based toxicity, including poor selectivity for the hERG channel, phototoxicity, and phospholipidosis. However, other inhibitors in clinical trials appear more promising [47]. Overall, SHP2's role in cancer plasticity makes targeted combination therapies with SHP2 inhibitors versatile, with many potential targets for combination therapy.
5 The Role of Phosphatases in Cell Crosstalk Through Exosome Secretion
Exosomes play a crucial role in tumor progression by upregulating PD-1 expression, promoting macrophage polarization, modulating cytotoxic T-cell activation, and establishing a pre-metastatic niche through enhanced cell crosstalk in the TME [28, 29, 31-33]. In addition to their intracellular role in tumor progression, phosphatases also influence the TME [28, 29, 33]. Notably, they have been implicated in regulating exosome secretion, which promotes tumor progression, as will be discussed in this section. Tumor-derived exosomes are formed through the establishment of a pre-metastatic niche, promoting tumor colonization and metastasis [211].
Exosomes promote immunosuppression through specific phosphatase overexpression [28, 30, 31]. For example, circular ubiquitin-specific protease-7 (CircUSP7) upregulates SHP2 expression by depleting miR-934, which functions to downregulate SHP2 in NSCLC [31]. Specifically, high levels of circUSP7 induced PD-1 immunotherapy resistance in NSCLC through the exosome circUSP7/miR-934/SHP2 axis [31]. This study also demonstrated that high circUSP7 levels are associated with CD8+ T-cell dysregulation and subsequent immunosuppression mediated by SHP2 [31]. Exosomal miR-138-5p secretion modulates macrophage polarization, promoting the anti-inflammatory M2 phenotype through SHP2 downregulation [29]. Phosphatase dysregulation has also been found to directly affect exosome secretion in the TIME, promoting immunosuppression and metastasis [32, 33]. For example, SHP2 reduces macrophage activation by negatively regulating small extracellular vesicle (sEV) secretion through the dephosphorylation of tyrosine 46 in syntenin, thereby decreasing sEV transfer and macrophage activation [32]. However, this effect was reversed by SHP2 inhibition, leading to increased tyrosine phosphorylation of syntenin and a higher number of sEV transfers, which in turn induced macrophage activation and a robust inflammatory response [32]. Similarly, another study demonstrated that SHP2 expression promoted the release of miR-138-5p exosomes that activate the PI3K/AKT pathway, resulting in the differentiation of anti-inflammatory (M2) macrophages and invasion of colorectal cancer (CRC) cells [33]. Notably, another study demonstrated that the coculture of melanoma exosomes containing both SHP2 protein and mRNA with T-cells suppressed antitumor immune activity, which was reversed by SHP2 inhibition [28]. Another study revealed that SHP2 binds to phosphorylated tyrosine 720 of PDGFRα and mediates increased extracellular vesicle (EV) release via the mTOR pathway, promoting hepatic stellate cell migration and fibrosis in mouse models [212]. These findings illustrate SHP2's role in the crosstalk between tumor and immune cells, highlighting its therapeutic potential in reducing therapy resistance and tumor aggressiveness in the TME [28, 33, 212].
Another example of PTP modulating cross-talk in the tumor microenvironment is PTP receptor type O (PTPRO), a tumor suppressor [30]. In contrast to SHP2, PTPRO-derived exosomes induced macrophage polarization to the M1 pro-inflammatory phenotype and inhibited breast cancer cell invasion and migration by inducing STAT3 and STAT6 activation in macrophages [30].
Furthermore, exosomal miR-133b has been reported to suppress breast cancer proliferation In Vitro and In Vivo by upregulating DUSP1 and was found at low levels in the serum of cancer patients [213]. This provides a new therapeutic approach to target miRNAs that dysregulate phosphatases in malignancies, potentially in combination with other therapies. Furthermore, PRL3 expression is also associated with increased exosome release, as tumor-derived exosomes containing PRL3 promoted colonization and metastasis in melanoma In Vivo models [198]. These findings highlight the critical role of phosphatases in cancer and their involvement in exosome-mediated tumor progression. However, further research is needed to fully elucidate the roles of phosphatases and exosomes in tumors and their potential as therapeutic targets.
6 Phosphatases as a Therapeutic Target in Clinical Trials
There are several ongoing clinical trials for phosphatases in patients with various malignancies. For example, AC484 targeting PTPN2 is in Phase 1 clinical trials as monotherapy and in combination with PD-1 blockade for solid tumors (ClinicalTrials.gov identifier NCT0477794). Another orally bioavailable PTPN2 inhibitor, ABBV-CLS-579, is in Phase I clinical trials for locally advanced or metastatic tumors (Table 1) (NCT04777994 and NCT04417465) (Table 2).
| Drug | Phosphatase target | Tumors | Clinical phase | Clinical trial | Companies | Status |
|---|---|---|---|---|---|---|
BBP-398
|
SHP2 | Advanced solid tumors including KRAS G12C NSCLC, NF1 loss of function and EGFR mutant NSCLC | Phase I/Ib | NCT04528836 | Navire Pharma Inc. | Ongoing |
| BPI-442096 | SHP2 | Advanced solid tumors, KRAS G12C NSCLC, class-3 BRAF, NF1 LOF mutations, and RTK mutations. | Phase I | NCT05369312 | Betta Pharmaceutical Co. Ltd | Ongoing |
| BR790 | SHP2 | Advanced solid tumors | Phase I | NCT04891653 | Jiangxi Qingfeng Pharmaceutical Co. Ltd. and Shanghai Gopherwood Biotech Co. Ltd. | Ongoing |
| ERAS-601 | SHP2 | Advanced or metastatic tumors with specific molecular alterations | Phase I/II | NCT04670679 | Erasca Inc. | Ongoing |
| ET0038 | SHP2 | Advanced solid tumors with specific mutations that drive RAS–MAPK hyperactivation | Phase I | NCT05354843 | Etern BioPharma (Shanghai) Co. Ltd. | Ongoing |
GDC-1971
|
SHP2 | Advanced or metastatic tumors excluding KRAS G12D, G12V,G13X, and Q61; BRAF V600; or mitogen-activated protein kinase kinase (MEK) mutations | Phase I/Ib | NCT04252339 | Genentech Inc./Relay Therapeutics | Ongoing |
| GH21 | SHP2 | Advanced or metastatic tumors | Phase I | NCT05183243 | Suzhou Genhouse Bio Co. Ltd. | Ongoing |
HBI-2376
|
SHP2 | Advanced malignant solid tumors harboring KRAS or EGFR mutations | Phase I | NCT05163028 | HUYABIO International LLC. | Ongoing |
| H5-10381 | SHP2 | Advanced solid tumors | Phase I | NCT05378178 | Jiangsu Hansoh Pharmaceutical Co. Ltd. | Ongoing |
| ICP-189 | SHP2 | Locally advanced unresectable or metastatic solid tumors | Phase I | NCT05370755 | Beijing InnoCare Pharma Tech Co. Ltd. | Ongoing |
JAB-3068
|
SHP2 | Non-small cell lung cancer Head and neck cancer Esophageal cancer |
Phase I/II | NCT03518554 | Jacobio Pharmaceuticals Co. Ltd. | Ongoing |
| JAB-3312 | SHP2 | Non-small cell lung cancer Colorectal cancer Pancreatic ductal cancer Esophageal squamous cell carcinoma Head and neck squamous cancer Breast cancer Other solid tumors |
Phase I/II | NCT04045496 | Jacobio Pharmaceuticals Co. Ltd. | Ongoing |
PF-07284892
|
SHP2 | ALK- or ROS1 positive non-small cell lung cancer (NSCL), colorectal cancer, or RAS-mutant, NF-1 mutant or BRAF class 3-mutant solid tumors | Phase I/IB | NCT04800822 | Pfizer Inc. | Ongoing |
RMC-4630
|
SHP2 | Advanced solid tumors, tumors with KRAS amplifications (NSCLC), BRAF Class 3, or NF1 LOF mutations in NSCLC and gynecological cancers | Phase I/II | NCT03634982 | Revolution Medicines Inc. | Ongoing |
| SH3809 | SHP2 | Advanced solid tumors (except HCC) | Phase I | NCT04843033 | Nanjing Sanhome Pharmaceutical, Co. Ltd. | Ongoing |
TN0155
|
SHP2 | Advanced solid tumors excluding known KRAS, NRAS, HRAS, BRAF, or PTPN11 mutations | Phase I/II | NCT03114319 | Novartis Pharmaceuticals | Ongoing |
ABBV-CLS-484
|
PTP1B/TCPTP | Locally advanced or metastatic solid tumors | Phase I | NCT04777994 | Calico Life Sciences LLC | Ongoing |
| PLR3-zumab | PRL3 | Gastric cancer, hepatoma cellular carcinoma, advanced solid tumors | Phase II | NCT04118114, NCT04452955 | Intra-ImmuSG Pte. Ltd | Ongoing |
MSI-1436C
|
PRL3 | Metastatic breast cancer | Phase I | NCT03191682 | Genaera | Terminated |
LB-100
|
PP2A and PP5 | Astrocytome (grade 2,3,4), glioblastoma multiforme, myelodysplastic syndrome | Phase II | NCT03027388 | LIXTE | Completed |

- Source: Larvol Clinical Trials Database. Accessed March 2025. https://clin.larvol.com/trial-detail/.
Preclinical studies demonstrated that PTPN2/N1 inhibition enhanced T-cell antitumor immunity by targeting tumor and immune cells within the TME [148]. Modulation of PTPN2 activity could provide a novel therapeutic strategy to improve the efficacy of cancer treatments, particularly in solid tumors where T-cell responses are suppressed (Table 1).
A PTP1B inhibitor, AC-484, was the first catalytic inhibitor to enter clinical trials for cancer immunotherapy [214]. AC-484 demonstrated high tolerability and efficacy in preclinical models, even in cases of PD-1 blockade resistance, which has implications for patients who exhibit resistance to PD-1 immune checkpoint therapies [173]. ABBV-CLS-484 exhibited dose-dependent inhibition of PTPN2/PTP1B and increased immune cell levels and infiltration in the tumor tissues of MC38 tumor-bearing mice (Table 1) [173].
Sodium stibogluconate (SSG), a catalytic SHP1 inhibitor, is highly effective in combination with interferon alpha-2b [215]. SSG has been used in Phase I clinical trials for malignant melanoma (NCT00498979) and advanced malignancies (NCT00629200) in combination with interferons. However, the trial was terminated owing to high toxicity effects, including thrombocytopenia, fever, chills, and pancreatitis [91]. Nonetheless, SHP1 remains a promising target for regulating oncogenic STAT3, which suppresses tumor growth, chemoresistance, and antitumor immune activity.
LB-100 is a small-molecule inhibitor targeting PP2A in clinical trials, and have been shown to overcome cisplatin resistance in medulloblastoma (Table 1) [216]. Furthermore, LB-100, in combination with temozolomide and doxorubicin, suppressed tumor growth in glioblastoma, pancreatic cancer, and hepatocellular carcinoma (HCC) [217, 218]. LB-100 has completed a Phase I clinical trial for treating solid tumors in combination with Docetaxel (NCT01837667) and is currently in Phase II clinical trials in recurrent glioblastoma patients (NCT03027388) and in patients with myelodysplastic syndromes (NCT03886662). Additionally, a Phase 1b clinical trial is underway in combination with chemotherapeutic agents in patients with advanced small cell lung cancer (NCT04560972).
A humanized antibody targeting PRL3, known as PRL-3-zumab, is currently in Phase I clinical trials for treating advanced solid tumors (Table 1) [134]. PRL-3-zumab inhibited tumor growth of PRL3+ gastric cancer xenograft models and prevented tumor recurrence after surgical resection, with minimal side effects [219]. In addition to inhibiting metastasis by targeting PRL3, PRL-3-zumab also enhanced the antitumor immune response by recruiting immune cells [134]. These findings highlight PRL3 as a promising therapeutic target for both immunotherapy and metastasis inhibition, with minimal adverse effects.
SHP2 inhibitors are under extensive investigation in clinical trials targeting advanced cancers. RMC-4630, a potent SHP2 inhibitor, was evaluated in Phase 1b/2 trials for malignancies with oncogenic mutations, such as BRAF G466V and G596R, demonstrating tumor growth inhibition and disruption of the RAS/RAF/MEK/ERK pathway (NCT03634982) [40]. In a Phase 1b trial, RMC-4630 was combined with Cobimetinib (a MEK inhibitor) to assess safety, pharmacokinetics/pharmacodynamics (PK/PD), and antitumor activity in patients with relapsed or refractory solid tumors harboring oncogenic mutations (NCT03989115) [40]. Another Phase 1b trial tested RMC-4630 with AMG510 (a KRAS G12C inhibitor) in advanced solid tumors with KRAS G12C mutations, focusing on tolerability, safety, and PK/PD (NCT04185883) [40].
TNO155 is also in clinical development. In a Phase 1/2 trial, it was combined with MRTX849 (a KRAS G12C inhibitor) to evaluate efficacy and safety in patients with KRAS G12C-mutant tumors (NCT04330664). A separate Phase 1 trial investigated TNO155 with Nazartinib (an EGFR inhibitor) to determine the safe dosage in RTK-dependent solid tumors, showing a synergistic effect in preclinical models of EGFR-mutant lung cancer (NCT03114319). TNO155 is also part of a Phase 1b trial with Spartalizumab (anti-PD-1 antibody) or Ribociclib (CDK4/6 inhibitor) to assess safety, tolerability, and antitumor activity in various malignancies, including RTK-dependent tumors (NCT04000529, NCT04699188).
Other SHP2 inhibitors include BBP-398, currently in a Phase 1 trial for solid tumors with RAS and RTK mutations, focusing on optimal dosage, objective response rates, and response duration (NCT04528836). RLY-1971 is being tested in a Phase 1 open-label study for advanced or metastatic solid tumors in combination with KRAS G12C inhibitors to determine the maximum tolerated dose, safety, and efficacy (NCT04252339). ERAS-601 advanced to Phase 1/1b trials as monotherapy or in combination with MEK inhibitors, aiming to evaluate safety, tolerability, and PK/PD in patients with advanced or metastatic solid tumors (NCT04670679) [40].
JAB-3068 entered Phase 1/2a trials in the USA and China to determine the effective dosage for patients with advanced solid tumors (NCT03518554, NCT03565003) and received FDA orphan drug designation for esophageal cancer therapy. JAB-3312 is in a Phase 1 trial assessing safety, efficacy, and tolerability in patients with advanced solid tumors (NCT04045496).
These trials aim to explore the safety, efficacy, and combinatorial potential of phosphatase drugs in various cancers, with the goal of overcoming resistance mechanisms and enhancing antitumor responses.
7 Conclusion and Future Perspectives
Cancer cells rely on phosphatase dysregulation for its survival through modulating proliferation or metastases [41, 51, 217]. These enzymes relay RAS/MAPK, PI3K/AKT, and JAK/STAT pathway signaling downstream of RTK or TCR, driving tumorigenesis and immunosuppression, respectively [13, 86, 88, 220, 221].
Depending on the context, some phosphatases can function either positive and negative modulator or both, depending on the cell type and the downstream substrates they target. For example, SHP2 negatively regulates immune cells while promoting tumorigenesis in cancer cells [43, 48, 222]. Furthermore, phosphatases exhibit overlapping functions in both tumor cells and immune cells, targeting multiple dysregulated phosphatases simultaneously could enhance antitumor activity and have synergistic effect. For instance, dual inhibitors like 1B/TC, which target PTP1B and PTPN2, have shown promise in preclinical studies [223]. However, phosphatases with dual functions in tumors must be targeted in a context-dependent manner to minimize off-target effects.
Specific phosphatases play a distinct role in cancer progression and immune regulation. For instance, PTPN22 and SHP2 regulate cytokine secretion and immune checkpoint blockade, whereas PRL3 and DUSP1/6 modulate key oncogenic pathways such as MAPK and PI3K/AKT [43, 85, 115]. Additionally, phosphatases like SHP2 and PP2A influence metastasis by modulating Hippo pathway regulation [224-226]. Both SHP2 and PRL3 also promote metastasis by enhancing chemokine receptor expression on cells, thereby recruiting cancer cells to distant sites [65, 227]. These findings underscore the potential of targeting these phosphatases to target intrinsic and extrinsic mechanisms to suppress tumor progression. SHP2 is the most well understood phosphatase and more needs to be elucidated on its mechanisms in cancer.
Phosphatases also play critical roles in immune cells and the broader TME, making them attractive targets for cancer immunotherapy. Studies have shown that targeting specific phosphatases (e.g., SHP2, PTPN22), either alone or in combination with chemo- or immunotherapy, provides synergistic antitumor immune activity in preclinical studies and clinical trials [13, 14, 41, 83, 200]. These effects include enhanced tumor cell clearance, a high CD8+ to Treg cell ratio, improved immune signaling, and increased sensitivity of tumor to pro-inflammatory cytokines [3, 9, 155, 176, 190]. Phosphatases also significantly influence tumor cell-TME crosstalk [228]. Tumor-derived exosomes can either inhibit or promote tumor progression by modulating phosphatase activity. Targeting phosphatase-regulating exosomal transcripts, such as circUSP7 and miR-138-5p, presents additional therapeutic opportunities [229, 230].
Targeting phosphatases alone and in combination with TKIs has shown strong efficacy In Vitro and In Vivo studies [22, 44, 46, 92]. However, despite the clinical advancement of SHP2 inhibitors, discrepancies between preclinical and clinical outcomes have hindered translational progress [231]. Advanced imaging techniques, such as fluorescence imaging, could help bridge this gap by providing deeper insight into drug mechanisms and interactions [232]. Emerging strategies—including PROTAC and biologics—offer innovative avenues for phosphatase-targeting therapies [134, 186, 233].
Among phosphatases, SHP2 is the most well characterized, with multiple inhibitors in clinical trials [40, 46, 208, 234-236]. However, the mechanisms of action of other phosphatases, such as PRL3, PTPN22, DUSP, remain less understood. While significant progress has been made in phosphatase-targeted therapy, a deeper understanding of their roles within the TME is essential to fully unlock their therapeutic potential in cancer treatment.
Author Contributions
Maryam Jama: Writing – original draft, writing – review & editing, conceptualization. Michael Overduin: Writing – review & editing. Khaled H. Barakat: Writing – review & editing. All authors have read and approved the final manuscript.
Acknowledgments
This study was supported by the Natural Sciences and Engineering Research Council of Canada (NSERC) Discovery Grant [RGPIN-2020-04437], the Natural Sciences and Engineering Research Council (RGPIN-2024-06426 and I2IPJ 548807-2020), and Canada Foundation for Innovation John Evans Leaders (38496) grants.
Ethics Statement
The authors have nothing to report.
Conflicts of Interest
The authors declare no conflicts of interest.
Open Research
Data Availability Statement
No new data were generated or analyzed in this review. All data used in this study were obtained from previously published articles and sources that are referenced throughout the manuscript.

