

p38 mitogen-activated protein kinase: Functions and targeted therapy in diseases
Qinwen Zheng, Shutong Li, and Aoxue Wang contributed equally to this study.
Abstract
P38 mitogen-activated protein kinase (p38MAPK) is a multifunctional protein kinase that plays an important role in human normal physiological activities and a variety of major diseases, and its signaling pathway affects a variety of regulatory factors in vivo, which is related to cell cycle, survival, metabolism, and differentiation. The four subtypes of p38MAPK have significant differences in their distribution, content, and effects in the body. The inhibitors of the four subtypes play potential roles in regulating cancer, neurodegenerative diseases, inflammation, and cardiovascular diseases, making it an attractive target for drug development. So far, an increasing number of p38MAPK inhibitors have been developed for targeted therapy in diseases, among which some representative compounds have entered clinical trials. Therefore, this review aims to provide a summary of the structural characteristics, signaling pathways of P38MAPK and the relationship between p38MAPK and disease, along with an overview of the binding modes and structure–activity relationships of small molecule inhibitors targeting four p38MAPK subtypes, and summarizes the challenges about the development of p38MAPK inhibitors, hoping to provide a valuable reference for the development and application of novel inhibitors.
Graphical Abstract
Our review provides an overview of the relationship between P38 mitogen-activated protein kinase (p38MAPK) and disease, especially on small-molecule inhibitors of four p38MAPK subtypes, and summarizes the challenges about the development of p38MAPK inhibitors.

1 INTRODUCTION
Mitogen-activated protein kinase (MAPK) is a group of serine-threonine protein kinases that can be activated by different extracellular stimuli. MAPK contains four subgroups: extracellular regulated protein kinases (ERK), p38 protein kinase (p38MAPK), c-Jun N-terminal kinase (JNK), and ERK5.1 Among them, p38MAPK was discovered by Brewster et al. in 1993 when studying the influence of hypertonic environment on fungi and later found in mammals. It was first identified as a 38 kDa protein, so it was called p38. P38MAPK consists of four subtypes: p38α (MAPK14), p38β (MAPK11), p38γ (MAPK12), and p38δ (MAPK13). Some opinions also believed that p38MAPK was composed of five subtypes, and p38β1 and p38β2 were subdivided.2 The four isomers exist in the nucleus and cytoplasm, among which p38α is the most widely distributed, followed by p38β, while p38γ and p38δ tend to be specifically expressed in specific organ cells, and they are concentrated in skeletal muscle and endocrine glands, respectively.3 The homology of the four different subtypes is also different, which is relatively simple at the domain level.
P38MAPK is mainly activated through the pathway of mitogen-activated protein kinase kinase kinase (MAP3K)—mitogen-activated protein kinase kinase (MAP2K)–MAPK, and signaling pathway is activated to regulate a variety of substrates, including protein kinases, transcription factors, and transcription regulators.4 This enables it to participate in a large number of normal physiological processes, such as cell cycle, cell metabolism, and cell differentiation, and to influence the course of major diseases, such as cancer, neurodegenerative diseases, inflammation, immunity, cardiovascular diseases, and respiratory diseases, and so on. Similarly, due to the different distribution of different subtypes, the diseases caused by functional abnormalities are also different. For example, p38 MAPK subgroup is the most involved in airway and lung inflammation underlying asthma and chronic obstructive pulmonary disease (COPD). In particular, several environmental agents including aeroallergens, cigarette smoke, airborne pollutants, viral and bacterial pathogens activate the p38ɑ isoform which in turn upregulates the expression of multiple proinflammatory cytokines and chemokines, as well as the production of some fibrogenic factors.5-7 The inhibition of p38MAPK has been proven to have a potential effect on the above diseases. In recent years, a considerable number of inhibitors targeting four different subtypes have entered clinical trials, and the related inhibitors have significant antitumor cell proliferation and anti-inflammatory effects.
From this, it can be seen that p38MAPK inhibitors have been widely studied and have achieved good results. However, there is currently no corresponding review that systematically summarizes the p38MAPK inhibitors that have been published and have entered clinical trials so as to enable researchers to understand the current research status in this field and provide clearer ideas and directions for future research. Therefore, in this review, we summarize the structure, signaling pathway and activation mechanism of p38MAPK, the relationship between p38MAPK and diseases, the research progress of different subtypes of small molecule inhibitors, and the selectivity of four different subtypes were emphatically introduced from the perspective of pharmacochemistry. In addition, in the review, we focused on summarizing the current challenges faced by p38MAPK inhibitors and providing some opinions and considerations for their future development. We hope that through our review, future researchers can have a clearer understanding of the current opportunities and challenges in this field and provide ideas for the development of new p38MAPK inhibitors in the future.
2 STRUCTURE AND SIGNAL PATHWAY OF P38MAPK
2.1 Introduction to p38 structure
The MAPK subfamilies identified so far include ERK1/2, JNK1/2/3, p38α/β/γ/δ, ERK3, ERK4, and ERK5. P38 is composed of four isoforms with good homology among them: p38β, p38γ, and p38δ share 75%, 62%, and 64% homology with p38α, and about 40%–50% homology with other members of the MAPK family (Figure 1A). MAPK has two phosphorylation sites separated by an amino acid, forming the tripeptidyl TXY, and MAPK is divided into different subfamilies according to the difference of X in the tripeptidyl, among which the tripeptidyl of p38MAPK is TGY.

P38α (MAPK14) is composed of 360 amino acids. Its domains include Protein Kinase Domain and TXY Motif. The Protein kinase domain occupies almost the entire p38α, maintaining the main kinase role of p38α. Amino acids 106–109 play an important role in inhibitor design: Thr106 is known as the “gatekeeper” of adjacent sites involving Glu71 and Asp168. Several classes of p38α inhibitors with double-ring skeletons are able to form hydrogen bonds with the amide NH of Met109.8 The TXY Motif formed at amino acids 180–182 is located in a region called the “activation ring,” and phosphorylation of residues Thr180 and Tyr182 results in the activation of p38α.9
P38β (MAPK11) has 75% homology with p38α, and its domain is not much different from p38α. It consists of 364 amino acids, including the Protein kinase domain, two Inhibitor-binding Region, and TXY motif, and two Disordered Region. One of the Inhibitor-binding Region contains the amino acid sequence Asp168-Phe169-Gly170. Some inhibitors induce the “DFG-out” conformation by binding Asp168-Phe169-Gly170 at the beginning of the activation ring. Researchers also divide inhibitors into two corresponding types according to the p38MAPK binding of different conformations (DFG-in and DFG-out).
P38γ (MAPK12) is composed of 367 amino acids, and Protein kinase Domain and TXY Motif comprise its domain. The sites of Thr106, His107, and Leu108 in p38α and p38β are Met106, Pro107, and Phe108 in their more distant congeners p38γ and p38δ. The absence of Inhibitor-binding Region shared by p38α and p38β may also be the reason for the limited number of p38γ and p38δ inhibitors.
P38δ (MAPK13) contains 365 amino acids and is composed of Protein kinase Domain and TXY Motif. It is noteworthy that the activation sites of p38α, p38β, and p38δ are the same at amino acid positions 180–182, while the activation sites of p38γ are 183–185.
2.2 Activation process of p38
MAP kinase kinase (MKK) activates MAPK through simultaneous phosphorylation of threonine (T) and tyrosine (Y). P38MAPK needs to be phosphorylated in a flexible ring called phosphorylation lip or activation loop through the dual phosphorylation in the ring sequence Thr-Gly-Tyr to complete its activation. This phosphorylation induces conformational recombination to relieve spatial obstruction and stabilizes the activation ring by opening and extending the conformation, thereby facilitating its binding to the substrate.
- (1)
Cascade of MAP3Ks, MAP2Ks, and MAPK signals. The most common is that growth factors, inflammation, and other external conditions stimulate the activation of MAP3Ks, and then activate MAP2Ks, leading to the activation of MAPK (Figure 1B).
MAP2Ks include MKK3, MKK4, and MKK6. MKK3 and MKK6 are the most important for MAPK activation. MKK3 can activate other three subtypes except p38β, but its splicing variant MKK3b can activate p38β.10 MKK6 activates all subtypes of p38. In addition, MKK3 and MKK6 are critical for regulating the phosphorylation of p38γ substrate human homolog of Drosophila disc large (hDlg).
Although MKK3 and MKK6 have many similarities, for example, both matter in the activation of p38α and p38β, there are also significant differences in the activation process of p38. Under normal conditions, activation of p38δ by UV radiation, hypertonic shock, anisomycin, or tumor necrosis factor-α (TNF-α) is typically mediated by MKK3, that is, MKK3 is the dominant activator of p38δ. MKK6 phosphorylates and activates p38β and p38γ, and only activates p38δ in the absence of MKK3. In addition, MKK6 is a major p38γ activator in response to TNF-α. Meanwhile, MKK3 and MKK6 are activated by different MAP3Ks.11
In addition, MKK4, as an activator of the JNK pathway, can also activate p38α. However, MKK4 has not been found to have significant effects on several other subtypes.
MAP2Ks are activated by MAP3Ks through phosphorylation of two conserved serine and threonine sites in the activation ring. For MAP3Ks, its regulation is more complex and depends more on different types of activation modes of specific cells. MAP3K, which is known to have an obvious activation effect on MAP2Ks, has apoptosis signal-regulating kinase 1(ASK1), delta-like noncanonical Notch ligand 1, transforming growth factor-β-activated kinase 1 (TAK1), thousand-and-one amino acid 1/2, tumor progression loci 2, mixed-lineage kinase 3, MAPK/ERK kinase kinase(MEKK3), MEKK4, leucine zipper, and sterile-α motif kinase 1. In addition, some MAP3K can also activate the JNK pathway.
The activator upstream of MAP3K is more complex, involving the phosphorylation of STE20 family kinases, the regulation of Rho family, Cdc42, Rac, TNFR-associated factor 2/3/6(TRAF2/TRAF3/TRAF6), and many other factors. It is precisely this complexity regulation that enables the p38MAPK pathway to integrate the influence of a variety of different excitation factors, which further proves the importance of the p38MAPK pathway.
- (2)
The second type is about T-lymphocyte stimulated by T-cell receptor (TCR). For example, tyrosine kinase Zeta-chain associated protein 70 near TCR and p56lck activate Tyr323 when phosphorylating p38α. This results in the activation of p38α autophosphorylation on the ring.12
- (3)
Another pathway involves TAK1-binding protein 1 (TAB1), which can bind to p38α, but not to other members of the p38MAPK family, and induce the autophosphorylation of p38α in the activation ring.13
- (4)
At last, Im et al. proposed a fourth atypical MAP2K independent mechanism, suggesting that p38 MAPK activation occurs through a unique signaling pathway induced by cell division cycle 7 depletion. This activation requires a major sensor kinase ataxia telangiectasia—mutated and Rad3-related kinase (ATR) for checkpoint response.14 However, the underlying mechanism remains unclear.
2.3 Substrates of p38
P38MAPK can be combined with a variety of downstream targets, including protein kinases, transcription factors, and transcription regulators, and so on thus activating downstream signaling pathways and affecting a variety of cell processes including survival, proliferation, differentiation, and apoptosis (Figure 1B).
P38α has the most substrates among the four subtypes and is the most widely studied subtype. It activates most of the p38MAPK protein kinases, these included MAPK-activated protein kinase (MK)2/3/5,15, 16 MAPKs-interacting protein kinases,17, 18 mitogen-and stress-activated kinase (MSK)1/2,19 p21-activated kinase 6,20 while inhibiting phosphatidylinositol 5 phosphate 4-kinase,21 glycogen synthase kinase (GSK)3β.22 In addition, p38α also enables Protein Kinase C epsilon (PKCε) to complete cytokinesis.23 These protein kinases are important in many regulations. For example, MK2 and MK3 are phosphorylated primarily by the AU-rich element (ARE) binding proteins tristetraprolin (TTP) and HuR, it is also involved in the control of gene expression at the posttranscriptional level by regulating eukaryotic elongation factor 2 kinase. MNK1 and MNK2 regulate protein synthesis by phosphorylation of an initiation factor eukaryotic initiation factor 4E.24 MSK1/2 can directly phosphorylate and activate some transcription factors, playing an important role in the early genes that induce chromatin remodeling under stress or mitosis.25
Targets of p38α also include transcription factors, which are important in metabolism and apoptosis. CCAAT/enhancer-binding protein α (C/EBPα), C/EBPβ, and C/EBPε are only substrates of p38α in the four isoforms. In a study of p38α in diabetic mice, Qiao et al.26 found that p38α activation stimulated C/EBPα phosphorylation and showed that C/EBPα mediated p38α-stimulated PEPCK transcription in hepatocytes, which has significant implications for the treatment of diabetes. P38α can increase the stability of p53 protein and thereby regulate apoptosis.27 In Michigan Cancer Foundation-7 cells, p38α kinase activates p53 more effectively than other members of the Ras pathway. The co-expression of p38MAPK stabilizes p53 protein. Inhibition of p38α activation after UV irradiation can reduce the phosphorylation of p53 at Ser33, Ser37, and Ser15, and significantly reduce UV-induced apoptosis in a p53-dependent manner.
In addition, several chromatin remodeling regulators have been targeted for p38α. For example, for BRG1/BRM-associated factor 60c (BAF60c) it promotes the combination of myogenic differentiation factor D (MyoD) with target genes, and labels chromatin, promoting signal-dependent recruitment of switching defective/sucrose nonferal (SWI/SNF) core to muscle genes, thereby promoting muscle formation. Phosphorylation of BAF60c at conserved threonine by activated p38α promotes signaling of MyoD-BAF60c incorporation into BrG1-based SWI/SNF complex, which remodels chromatin and activates transcription of MyoD target genes.28
There are many other types of p38α targets, some of which are involved in cell cycle regulation, such as Cdc25A29/B,30 CyclinD131/3,32 and so on. It is involved in regulating cell death, such as Bax,33 BimEL,34 and so on. DNA/RNA binding proteins, such as chromatin licensing and DNA replication factor 1,35 Drosha,36 far upstream binding protein37; Endocytosis regulator, such as Early Endosomal Antigen 1,38 Rabenosyn-5 et al. Interacting MAPK pathway modulators, such as JNK-interacting protein 4,39 TAB1.40 Moreover, membrane protein epidermal growth factor receptor,41 fibroblast growth factor receptor 1,42 and structural protein heat shock (Hsp)27 are involved,43 Tau,44 and so on.
The downstream target of p38β was significantly less than that of p38α. Due to its high homology with p38α, it has several downstream protein kinases that are identical with p38α, such as MK2/3/5 and PKCε.
However, most of the other downstream targets are the same as p38α, only a few targets are specific targets of p38β, such as GS and Raptor. In a related experiment, Kuma et al.45 demonstrated that endogenous GSs in mouse skeletal muscle, liver, and brain extracts specifically bind to stress-activated protein kinase (SAPK) 2b/p38β, but not other members of the SAPK/p38 kinase group. This suggests that p38β may be a priming kinase that allows glycogen synthetase kinase 3 to phosphorylate Ser640, thereby inhibiting glycogen synthetase activity. Another specific substrate is Raptor, a regulatory subunit of mammalian target of rapamycin complex 1 (mTORC1), whose key phosphorylation sites are Ser863 and Ser771. Wu et al.46 demonstrated that the p38 pathway can regulate mTORC1 and thus affect autophagy. P38β can target different substrates to positively or negatively regulate mTORC1 activation when cells encounter different environmental stresses.
P38γ and p38δ also largely share the same substrate because of their homology. As subtypes of p38, both of them also have MK247, 48/MK3,15 two common substrates. In addition, MAF bZIP transcription factor A (MafA) can be used as substrate for p38γ and p38δ,49 which can enhance the transcription activity of MafA after binding. CyclinD3,32 Tau44 and DEP-domain-containing mTOR-interacting protein50 are also substrates for p38γ and p38δ isoforms.
In particular, c-JUN can be used as a substrate of p38α, p38β, p38γ instead of p38δ, while myocyte enhancer factor 2A can be used as a substrate of p38α, p38β, p38δ instead of p38γ.51 It also reflects the certain specificity of p38γ and p38δ. P38γ-specific substrates include synapse-associated protein 97 (SAP97), a scaffold protein that forms polyprotein complexes with various proteins. Cytoskeleton is targeted by binding to guanylate kinase-associated protein (GKAP). Sabio et al.52 demonstrated that p38γ catalyzed SAP97/hDlg phosphorylation to trigger its separation from GKAP, thereby releasing it from the cytoskeleton. This may regulate the integrity of intercellular junction complexes, as well as cell shape and volume in response to osmotic stress. In 1998, researchers had found that Stathmin is a specific substrate for p38δ,53 the sites of phosphorylation were mapped to Ser-25 and Ser-38, and they also showed that p38δ phosphorylated Stathmin more efficiently than other p38 isoforms in vitro, but its specific physiological effects remain unclear.
3 PHYSIOLOGICAL ROLES AND FUNCTIONS OF P38MAPK
3.1 Normal physiological activity of p38MAPK
3.1.1 Embryo development function of p38
P38MAPK is important in the regulation of inner cell mass cells, especially in the differentiation of primitive endoderm. It is closely related to ribosome-related gene expression, rRNA precursor processing, polymer formation and protein translation regulation. Bora et al.54 found that DDX21 protein was dependent on active p38MAPK for rRNA processing regulation, and became the only nucleolus during blastocyst maturation. In addition, studies have shown that p38MAPK provides a permissible translation environment during the predifferentiation of mouse blastocysts.55
3.1.2 Immune response function of p38
T lymphocytes are the most important part in the immune response, and they are activated by TCR, co-stimulatory signaling, and immunomodulatory cytokines such as Interleukin (IL)-1/2 and IL-4. P38-MSK1/2-cAMP response element-binding protein/activating transcription factor 1 (ATF1) signaling pathways can be used to restrict Toll-like receptors-induced inflammation.56 In addition, many cytokines such as IL-12 and IL-4 are regulated by p38MAPK. When Maeyer et al.57 studied the immune response of the elderly; it was found that the decrease of T-cell immunoglobulin and mucin domain containing 4 (TIM-4) in the elderly was caused by the increase of p38MAPK activity in macrophages. Oral administration of active p38 inhibitors in older adults can reduce TIM-4 reduction. This suggests that p38MAPK plays an important role in cellular immune response.
3.1.3 Cell cycle function of p38
The p38 signaling pathway regulates the cell cycle by influencing the timing of cell cycle entry and cell cycle checkpoint stop. However, p38 plays a more important role in the regulation of inflammation, so as a part of cellular stress response, it tends to act as a “brake” to inhibit cell cycle transition.58 However, Whitaker et al.59 confirmed that p38 affected G1 and G2 phases. The tumor suppressor mechanism for G1 mediated by p38 includes cyclin-dependent kinases (CDK)—retinoblastoma suppressor protein—Early 2 factor pathway phosphorylation of several proteins, stress-activated p38 also prevents G2 phase cell transition to M phase cell cycle through the checkpoint mechanism of G2 phase, leading to G2 phase cycle arrest.
3.1.4 Cell differentiation function of p38
Multiple experiments have shown that p38MAPK also plays an important role in cell differentiation, for example, Weng et al.60 demonstrated that amiodarone regulates cell proliferation and myofibroblasts of human embryonic lungfibroblasts by regulating ERK1/2 and p38MAPK pathways. Kang et al.61 demonstrated that cannabinol regulates osteoblast differentiation in U2OS and MG-63 by enhancing protein–protein interactions between runt-related protein 2, Osterix, or phosphorylated p38MAPK.
3.1.5 Cell metabolism function of p38
Mesenchymal stem cell (MSC) plays a great role in regenerative medicine, organ transplantation, and other fields, while stress-induced p38MAPK signal has been proved to damage the function of MSC. Budgude et al.62 found that drug inhibition of p38MAPK could restore the vitality of MSC cells and enhance their function.
3.1.6 Cell senescence function of p38
Many previous experiments have confirmed that p38MAPK is related to the aging of skeletal muscle and other tissues. He et al.63 demonstrated that intestinal stem cell senescence is driven by mTORC1 via the p38MAPK-p53 pathway. Tao et al.64 demonstrated that overexpression of forkhead box protein A2 reduces cigarette smoke-induced cellular senescence and lung inflammation by inhibiting the p38 and Erk1/2 MAPK pathways.
3.1.7 Cell survival and death function of p38
Shi et al.65 studied the effect of phosphatase of regenerating liver-3 (PRL-3) on cell survival, and proved that PRL-3 inhibits phosphorylation by binding with p38MAPK, thus promoting cell survival. However, Filomeni et al.66 demonstrated that disulfide stress agents trigger cell death through REDOX activation of Thioredoxin 1/p38MAPK/p53 dependent signaling cascade.
3.2 Connection among p38MAPK and diseases
3.2.1 Connection between p38MAPK and cancer
The p38MAPK signaling pathway is commonly known as the SAPK pathway, which means that it is important in many diseases and strongly associated with cancer. As mentioned above, p38MAPK is associated with cell cycle, differentiation, metabolism, senescence, and other cellular processes, while cancer cells can disrupt pathways to promote proliferation, survival, and invasion. The p38MAPK pathway regulates cell cycle progression and cellular processes of cell survival and differentiation at different transition points through transcription-dependent and transcription-independent mechanisms,67 resulting in its influence on a variety of cancers.
In many cancer cells, such as liver cancer and lung cancer, p38α activation is usually associated with antiproliferation function, and p38α negatively regulates abnormal proliferation in various types of primary cells, including cardiomyocytes, hepatocytes, fibroblasts, hematopoietic cells, and lung cells.68, 69 P38α also plays a proapoptotic role in some cells. In the early transformation process, reactive oxygen species (ROS) will induce the activation of p38α to induce apoptosis, thus preventing the carcinogenic effect generated by the accumulation of ROS.70 P38α also mediates cell survival through a quiescent state called cancer dormancy, which is important for drug resistance in cancer cells. P38α is associated with G2/M checkpoint, which induces cell cycle arrest and promotes DNA repair, possibly leading to apoptotic resistance in cancer cells.58
P38β is thought to have antiapoptotic effects in various cell lines and may counteract the proapoptotic effects of p38α-activated cells.67 But beyond that, it also has the ability to regulate transforming growth factor (TGF) -1β and vascular growth factor-mediated endothelial cell survival in the absence of proapoptotic p38α.71 In addition, p38β is important in the regulation of oncogene Lipocalin 2 (LCN2), which is a target gene of plakophilin 3 (PKP3). When PKP3 expression is decreased, p38β can regulate LCN2, leading to increased tumor invasion and metastasis.72 Recent studies have detected elevated levels of p38β in a variety of female cancers, including breast cancer and endometrial cancer, suggesting that p38β may play a potential role.73
P38γ modulates gamma radiation-induced G2 phase arrest.74 In addition, p38γ acts as a CDK kinase, synergistic with CDK to regulate the G0 to G1 transition into the cell cycle. In cancer cell therapy studies, the absence of p38γ or treatment with p38γ inhibitors was found to prevent chemically-induced hepatocellular carcinoma formation, and biopsies of human hepatocellular carcinoma showed high expression of p38γ,75 demonstrating that p38 regulates some cancer cell formation.
P38δ is important for the movement and invasion of bile duct cancer cells, suggesting that it may play an important role in the metastasis of bile duct cancer.76 P38δ also has a regulatory effect on tumor genesis, p38δ-deficient mice showed decreased susceptibility to dimethylbenz (a) anthracen and 12-O-tetradeca-noylphorbol-13-acetate for treaty-induced skin cancer and Kras-induced lung cancer.77
In summary, p38MAPK is associated with many types of cancer. For example, in head and neck squamous cell carcinoma, activation of p38MAPK plays a role in treatment resistance and disease recurrence by maintaining tumor stem cell phenotype.78 In breast cancer, microRNA-3188 (miR-3188) can affect the proliferation, apoptosis, and migration of breast cancer cells by activating p38MAPK signaling pathway by targeting tumor suppressor candidate 5.79 In liver cancer, exposure to benzo (a) pyrene (BaP) can promote migration and invasion of HepG2 cells, which can be blocked by p38MAPK inhibitors, demonstrating that p38MAPK pathway is involved in Bap-induced migration and invasion of liver cancer cells.
3.2.2 Connection between p38MAPK and neurodegenerative disease
Strict control of the MAPK signaling pathway has been associated with many neurodegenerative diseases, including Alzheimer's disease (AD), Parkinson's disease (PD), Amyotrophic lateral sclerosis (ALS), and the activation of p38MAPK mediates neuronal apoptosis in AD, PD, and ALS.
AD is thought to be the formation of senile plaques containing amyloid-beta (Aβ) and neurofibrillary tangles containing the microtubule-associated protein tau in the brain.80, 81 Previous studies have shown that82 amyloid precursor protein dimerization mediated by Aβ42 can induce the activation of ASK1-MKK6-p38 signaling pathway, thus leading to tau phosphorylation. In addition, oxidative stress triggered by ROS is also an important factor in the pathogenesis of AD, and ROS is also a typical activator of p38MAPK in AD.83, 84 In the study of Mohamed et al.,85 it was demonstrated that sesame oil may regulate different molecular targets involved in the pathogenesis of AD by altering p38MAPK signaling.
PD is thought to be a disease caused by the gradual loss of dopaminergic neurons in the substantia nigra, and the accumulation of Lewy bodies (LBs) in the brain, in which specific proteins including modified alpha-synaptic nucleoproteins are deposited. Among them, alpha-synaptic nucleoprotein is present in LBs in the brain of patients with PD and plays a key role in the development of PD.86 The increased level of α-synaptonucleoprotein is thought to be associated with neuronal apoptosis induced by dysfunction of MAPK signaling pathway. Among them, alpha-synaptic nucleoproteins activate the p38, ERK, and JNK pathways in human microglia, leading to IL-1β and TNF-α production, which promote inflammation.87 In addition, p38MAPK and CD200-CD200R signals can regulate microglial cell dynamics in PD brain, and PD-related neurodegeneration is likely to be related to the destruction of CD200-CD200R and p38MAPK signal axes.88 In addition, p38 can phosphorylate and activate tumor suppressor p53, and then induce the expression of proapoptotic protein Bax, thereby inducing the permeability of the mitochondrial outer membrane.89 In addition, Obergasteiger et al.90 also proposed a new hypothesis that GTPase-p38 is used as upstream signal to treat PD through autophagy mechanism, which has certain potential possibilities.
Most cases of ALS occur by chance as a result of familial mutations in the Cu/Zn superoxide dismutase 1 (SOD 1) gene. Abnormal expression and activation of p38MAPK in motor neurons and microglia cells is considered to be an important factor in the pathogenesis of ALS.91 In addition, studies have shown that p38MAPK inhibitors can prevent motor neuron apoptosis induced by mutant SOD1.92 P38 and JNK1 are also associated with abnormal cytoskeleton of spinal motor neurons, and their abnormal phosphorylation and subsequent nerve filament aggregation are characteristic of familial and incidental ALS.93, 94
3.2.3 Connection between p38MAPK and inflammation
P38MAPK pathway is important in proinflammatory cytokines such as TNF-α, IL-1, IL-2, IL-6, IL-7, and so on,95 and the four subtypes of p38MAPK are also differentially expressed and activated in different inflammatory cells.96 In monocytes, p38α was the dominant expression form, but p38δ expression was low and p38β was not detected. The expression of p38α and p38δ was abundant in macrophages, but p38β was not detected. P38α and p38δ were significantly expressed in neutrophils, CD4+T cells and endothelial cells, while p38β was abundant in endothelial cells. P38γ was not detected in any inflammatory cells.
In studies of asthma, p38MAPK inhibitors have been shown to be effective in many in vitro and in vivo models of inflammation and help to resolve or regulate diseases such as asthma.97 Phosphorylated p38MAPK coordinates the phosphorylation of MSK1, and directly phosphorylates nuclear factor-kappa B (NF-κB) to produce type 2 cytokines. However, blocking NF-κB and p38MAPK with 4-carvomenthenol has been shown to improve combined allergic rhinitis and asthma syndrome.98
In studies of rheumatoid arthritis (RA), the pathological cause is believed to be the presence of a large number of proinflammatory cytokines in synovial fluid. It is later considered that the combination and neutralization of TNF-α and IL-1 proinflammatory cytokines are very effective for the treatment of RA. Preclinical models have shown that small molecule inhibitors have therapeutic potential.99, 100 P38α/β inhibitors were effective in a mouse model of collagen-induced arthritis, demonstrating that this class of p38MAPK inhibitor compounds prevents disease progression.101
3.2.4 Connection between p38MAPK and cardiovascular disease
P38MAPK also plays a role in cardiovascular disease, which is the leading cause of death worldwide. Previous studies have shown that the absence of p38α (MAPK14) in vascular smooth muscle cells can inhibit the formation of angiotensin-II-induced abdominal aortic aneurysm, vascular inflammation, and expression of aging markers.102 Atherosclerosis (AS) is also an important cause of myocardial infarction, coronary heart disease, and other heart diseases. Studies have shown that miR-124-3p overexpression can down-regulate the expression of MEKK3, inhibit p38MAPK signaling pathway, and thus inhibit macrophage proliferation in coronary AS mice. Promote macrophage apoptosis.102 Studies on diabetic cardiomyopathy (DCP) show that inflammatory factors influence the pathogenesis of DCP, and the signal transduction of NF-κB and p38MAPK is confirmed to be involved in the inflammatory response. Sirtuin-1 activation improves DCP by inhibiting phosphorylated p38MAPK in cytoplasm and NF-κB expression in nucleus.103
4 P38MAPK SMALL MOLECULE INHIBITORS
P38MAPK has been proven to play a role in a variety of major diseases, so the development of its inhibitors is more important. In the years of research and development of p38MAPK inhibitors, although no inhibitors have been approved for market, a considerable number of effective and selective inhibitors have been produced and shown good effects in preclinical and clinical trials. In this section, we summarized the research status of subtypes of classical inhibitors and novel inhibitors developed currently of p38 MAPK, hoping to provide a reference for the development p38MAPK inhibitors.
4.1 Inhibitors of p38α and p38β
P38α and p38β are the most widely effective subtypes of p38 in vivo, and many inhibitors targeting p38α/β have been widely developed. In recent years, the in-depth study of p38α has led researchers to develop selective inhibitors of p38α, which is widely used in the treatment of diseases such as RA.
4.1.1 Monocyclic p38α and p38β inhibitors
Imidazole derivatives
The pyridinyl imidazole compounds are considered to be the most effective inhibitors of p38α structure, and a number of selective inhibitors of p38α and p38β have been derived from these frameworks (Table 1).
| Inhibitor | Chemical name | Chemical structure | Activity | DMPK profile | References |
|---|---|---|---|---|---|
| Compound 1 (SB-203580) | 4-(4-uorophenyl)-2-(4-methylsulphinylphenyl)-5-(4-pyridyl)imidazole | 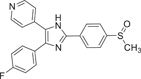 |
p38α IC50 = 0.1 μM p38β IC50 = 0.43 μM |
(mouse) Cm = 587 ± 173 ng·mL−1 Tm = 30 min Cl = 138 ± 39 mL·min−1·kg−1 |
[104-109] |
| Compound 2 (VRT-19911) | 4-(4-(4-fluorophenyl)-1-(piperidin-4-yl)-1H-imidazol-5-yl)pyridine | 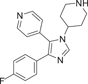 |
p38α Ki = 60 nM | Not tested | [110] |
| Compound 3 (CBS-3595) | N‑{4-[5-(4-Fluorophenyl)-3-methyl-2-methylsulfanyl‑3H‑imidazol-4-yl]-pyridin-2-yl}-acetamide | 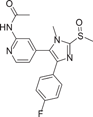 |
p38α IC50 = 0.5 μM PDE-4 IC50 = 0.2 μM |
Tm = 2.20 ± 1.30 h Cm = 0.20 ± 0.06 μM AUC0 - t = 0.95 ± 0.31 μM·h T1/2 = 2.7 ± 0.5 h |
[110, 111] |
| Compound 4 (ML3403) | {4-[5-(4-fluorophenyl)-2-methylsulfanyl-3H-imidazol-4-yl]-pyridin-2-yl}-(1-phenylethyl)-amine | 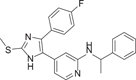 |
p38α IC50 = 0.38 μM, TNF-α IC50 = 2.7 ± 0.3 μM IL-1β IC50 = 0.99 ± 0.46 μM |
(Female) Tm = 1 h Cm = 386 ng/mL AUC0 - t = 1479 ng·h/mL |
[112, 113] |
| Compound 5 (SB242235) | 1-(4-piperidinyl)-4-(4-fluorophenyl)-5-(2-methoxy-4-pyrimidinyl) imidazole | 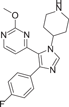 |
p38α IC50 = 19 nM p38β IC50 = 1 μM |
Cm = 280 ± 31 ng·mL−1 Tm = 60 min Cl =56 ± 8.9 mL·min−1·kg−1 T1/2 = 93.5 ± 11.3 min |
[114-117] |
| Compound 6 (RWJ67657) | 4-[4-(4-Fluorophenyl)-1-(3-phenylpropyl)-5-(4-pyrindinyl)-1H-imidazol-2-yl]-3-butyn-1ol | 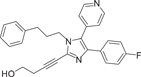 |
p38α IC50 = 1000 nM TNF-α IC50 = 3 nM IL-1β IC50 = 11 nM IL-8 IC50 = 0.04 μM |
(Female) Tm = 0.6 h T1/2 = 6.1 h F = 11% |
[118-120] |
| Compound 7 | 3-(2-phenyl-5-(2-((3-((phenylthio)amino)propyl)amino)pyridin-4-yl)-1H-imidazol-4-yl)phenol | 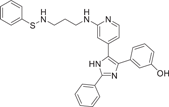 |
p38α IC50 = 47 nM TNF-α IC50 = 98.02 nM |
Not tested | [121] |
| Compound 8 | 3-(5-(2-((3-(((4-fluorophenyl)thio)amino)propyl)amino)pyridin-4-yl)-2-phenyl-1H-imidazol-4-yl)phenol | 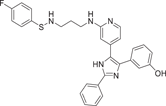 |
p38α IC50 = 45 nM TNF-α IC50 = 78.03 nM IL-6 IC50 = 17.6 µM IL-1β IC50 = 82.15 nM |
Not tested | [121] |
| Compound 9 (SC74102) | 4-(5-(4-fluorophenyl)-2H-imidazol-4-yl)pyridine | 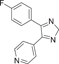 |
p38α IC50 = 600 nM | Not tested | [122, 123] |
| Compound 10 | 4-(3-(4-fluorophenyl)-5-(piperidin-4-yl)-1H-pyrazol-4-yl)pyridine | 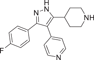 |
p38α IC50 = 25.3 nM p38β IC50 = 510 nM |
Not tested | [122, 123] |
| Compound 11 | 4-(3-(4-chlorophenyl)-5-(1-methylpiperidin-4-yl)-1H-pyrazol-4-yl)pyridine | 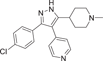 |
p38α IC50 = 31.6 nM p38β IC50 = 2680 nM |
Not tested | [122, 123] |
| Compound 12 (SC79659) | 1-(4-(3-(4-chlorophenyl)-4-(pyridin-4-yl)-1H-pyrazol-5-yl)piperidin-1-yl)-2-hydroxyethan-1-one | 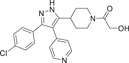 |
p38α IC50 = 111 nM p38β IC50 = 1790 nM |
Not tested | [122, 123] |
| Compound 13 (SD0006) | 1-(4-(3-(4-chlorophenyl)-4-(pyrimidin-4-yl)-1H-pyrazol-5-yl)piperidin-1-yl)-2-hydroxyethan-1-one | 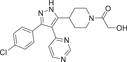 |
p38α IC50 = 110 nM p38β IC50 = 3450 nM |
Not tested | [122, 123] |
| Compound 14 | (5-amino-1-phenyl-1H-pyrazol-4-yl)(phenyl)methanone | 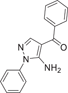 |
p38α IC50 = 2.3 μM | Not tested | [124, 125] |
| Compound 15 | (5-amino-1-(4-fluorophenyl)-1H-pyrazol-4-yl)(phenyl)methanone | 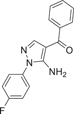 |
p38α IC50 = 1.1 μM | Not tested | [124, 125] |
| Compound 16 (RO3201195) | (S)-(5-amino-1-(4-fluorophenyl)-1H-pyrazol-4-yl)(3-(2,3-dihydroxypropoxy)phenyl)methanone | 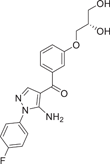 |
p38α IC50 = 0.7 μM | Not tested | [124, 125] |
| Compound 17 (DP-802) | 2-(3-(3-(tert-butyl)-5-(3-(2,3-dichlorophenyl)ureido)-1H-pyrazol-1-yl)phenyl)acetamide | 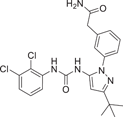 |
p38α IC50 = 9 nM BRAF IC50 = 17 nM CRAF IC50 = 11 nM |
Not tested | [126] |
| Compound 18 (SC409) | 4-(3-(4-chlorophenyl)-5-(1-methylpiperidin-4-yl)-1H-pyrazol-4-yl)pyrimidine | 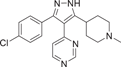 |
p38α IC50 = 0.04 μM p38β IC50 = 2.30 μM |
Not tested | |
| Compound 19 (TAK-715) | N-(4-(2-ethyl-4-(m-tolyl)thiazol-5-yl)pyridin-2-yl)benzamide | 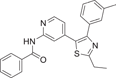 |
p38α IC50 = 240 nM TNF-α IC50 = 240 nM |
Not tested | [127] |
| Compound 20 (BMS-64099) | 2-(sec-butylamino)-N-(2-methyl-5-(methylcarbamoyl)phenyl)thiazole-5-carboxamide | 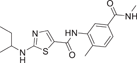 |
p38α IC50 = 3.5 nM TNF-α IC50 = 2.9 nM Lck IC50 = 22 μM |
Not tested | [128] |
| Compound 21 | 3-(2,5-dimethoxyphenyl)-N-(4-(4-(4-fluorophenyl)-2-(methylthio)-1H-imidazol-5-yl)pyridin-2-yl)propanamide | 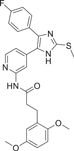 |
p38α IC50 = 10 nM | Not tested | [129] |
| Compound 22 | (E)-3-(2,5-dimethoxyphenyl)-N-(4-(4-(4-fluorophenyl)-2-(phenyldiazenyl)thiazol-5-yl)pyridin-2-yl)propanamide | 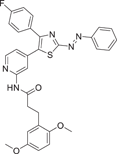 |
p38α IC50 = 2.36 ± 0.61 nM | Not tested | [129] |
| Compound 23 | 3-bromo-4-((2,4-difluorobenzyl)oxy)-6-methyl-1-phenylpyridin-2(1H)-one | 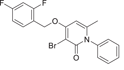 |
[130, 131] | ||
| Compound 24 | 3-bromo-4-((2,4-difluorobenzyl)oxy)-6-methyl-1-(o-tolyl)pyridin-2(1H)-one | 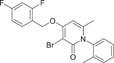 |
p38α IC50 = 12 nM | Not tested | [130, 131] |
| Compound 25 | 3-(3-bromo-4-((2,4-difluorobenzyl)oxy)-6-methyl-2-oxopyridin-1(2H)-yl)-N,4-dimethylbenzamide | 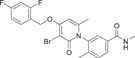 |
p38α IC50 = 2.3 nM | Not tested | [130, 131] |
| Compound 26 | 3-(3-bromo-4-((2,4-difluorobenzyl)oxy)-6-methyl-2-oxopyridin-1(2H)-yl)-4-fluoro-N-methylbenzamide | 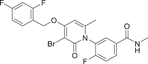 |
p38α IC50 = 8.5 nM | Not tested | [130, 131] |
| Compound 27 | 3-(3-bromo-4-((2,4-difluorobenzyl)oxy)-6-methyl-2-oxopyridin-1(2H)-yl)-4-chloro-N-methylbenzamide | 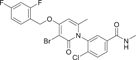 |
p38α IC50 = 3.0 nM | Not tested | [130, 131] |
| Compound 28 | 3-(3-bromo-4-((2,4-difluorobenzyl)oxy)-6-methyl-2-oxopyridin-1(2H)-yl)-4-(hydroxymethyl)-N-methylbenzamide | 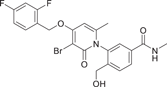 |
p38α IC50 = 325 nM | Not tested | [130, 131] |
| Compound 29 (Losmapimod) | 6-(5-(cyclopropylcarbamoyl)-3-fluoro-2-methylphenyl)-N-neopentylnicotinamide | 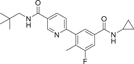 |
p38α pka = 8.1 p38β pka = 7.6 |
Not tested | [132-134] |
| Compound 30 (VX-702) | 2-(2,4-difluorophenyl)-6-(1-(2,6-difluorophenyl)ureido)nicotinamide | 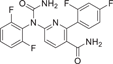 |
p38α IC50 = 4 ~ 20 nM | Not tested | [135, 136] |
| Compound 31 (UM-164) | N-(5-(3-(tert-butyl)benzamido)-2-methylphenyl)-2-((6-(4-(2-hydroxyethyl)piperazin-1-yl)-2-methylpyrimidin-4-yl)amino)thiazole-5-carboxamide | 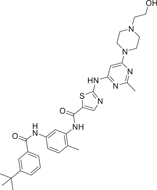 |
p38α Kd = 2.2 nM p38β Kd = 5.5 nM Src Kd = 2.7 nM |
Not tested | [137] |
| Compound 32 (UM101) | 4-chloro-N-(4-((1,1-dioxidothiomorpholino)methyl)phenyl)benzamide | 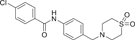 |
[138] | ||
| Compound 33 (PNU-120596) | 1-(5-chloro-2,4-dimethoxyphenyl)-3-(5-methylisoxazol-3-yl)urea | 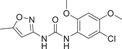 |
p38α IC50 = 3.3 μM | Not tested | [139, 140] |
| Compound 34 (VPC00628) | (R)-5-amino-N-(4-((1-amino-4-cyclohexyl-1-oxobutan-2-yl)carbamoyl)benzyl)-1-phenyl-1H-pyrazole-4-carboxamide | 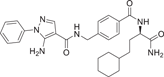 |
p38α IC50 = 7 ± 0.9 nM | Not tested | [141] |
| Compound 35 (VPC00630) | 5-amino-N-(((1 S,4r)-4-(((S)-1-amino-4-cyclohexyl-1-oxobutan-2-yl)carbamoyl)cyclohexyl)methyl)-1-phenyl-1H-pyrazole-4-carboxamide | 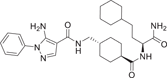 |
p38α IC50 = 44 ± 4.4 nM | Not tested | [141] |
| Compound 36 (VPC00257) | (S)-5-amino-N-(4-((1-amino-1-oxo-3-(3-(trifluoromethyl)phenyl)propan-2-yl)carbamoyl)benzyl)-1-phenyl-1H-pyrazole-4-carboxamide |  |
p38α IC50 = 46 ± 2.5 nM | Not tested | [141] |
| Compound 37 | (S)-5-amino-N-(4-((1-amino-1-oxo-3-(3-(trifluoromethyl)phenyl)propan-2-yl)carbamoyl)benzyl)-1-phenyl-1H-pyrazole-4-carboxamide |  |
p38α IC50 = 14.0 nM p38β IC50 = 16.8 nM |
Not tested | [142] |
| Compound 38 | N-(2-chloro-6-fluorobenzyl)-3-(furan-2-yl)-1H-1,2,4-triazol-5-amine |  |
Not tested | Not tested | [143] |
- Abbreviations: BRAF, B-RAF oncogene serine/threonine kinase; CRAF, C-RAF oncogene serine/threonine kinase; IL-1β, interleukin-1β; TNF-α, tumor necrosis factor- α.
Since it was developed by Gallagher et al. in 1995, SB-203580 (1) has been widely used as a potent and selective p38α/β inhibitor. As a pharmacological tool, it has been widely used to analyze the role of p38MAPK in various physiological processes.104 Compound 1 is a competitive inhibitor of ATP. The IC50 values of p38α and p38β are 0.1 and 0.43 μM, respectively, while the IC50 values of p38γ and p38δ are not inhibited. Kumar et al. also demonstrated that it only inhibited the activity of p38 mitogen-activated protein kinase, but not its activation.105 P38MAPK plays a role in Dengue virus (DENV)-induced hepatocyte apoptosis, and DENV infection induces p38MAPK phosphorylation. Its downstream signals include MAPKAPK2, HSP27, and AMP-dependent transcription factor-2 (ATF-2). Limjindaporn's team106 demonstrated that treatment with SB203580 reduced cytokines such as TNF-α, IL-6, and IL-10 in DENV infection, as well as regulated upon activation, normal T-cell expressed and secreted and interferon-gamma-inducible protein 10 were increased. Compound 1 did not reduce the phosphorylation of p38MAPK, but significantly reduced the phosphorylation of MAPKAPK2, HSP27, and ATF-2. It was confirmed that SB203580 modulated the downstream signal of p38MAPK to reduce DENV-induced liver injury. In addition, SB-203580 has been used to evaluate a wide range of phenomena such as bacterial sepsis,107 central nervous system (CNS) trauma response,108 and cardiac ischemia/reperfusion injury.109 As an earlier inhibitor of pyridinyl imidazole compounds, SB-203580 has derived a considerable number of other inhibitors with similar structure. Another inhibitor with similar structure belonging to pyridinyl imidazole compounds is VRT19911 (2). VRT19911 has been shown to bind to the ATP-binding site of p38α and is a selectively effective p38α inhibitor with a Ki of 60 nM. However, it has previously been reported that the presence of pyridinyl components in SB203580 and VRT19911 results in significant inhibition of hepatocytochrome p450 isoenzymes in vitro,144 and because oral activity is also reduced, these compounds are not suitable for the treatment of chronic diseases.
Albrecht et al.110 coincidentally discovered a dual-targeted p38αMAPK/Phosphodiesterase-4 (PDE-4) inhibitor CBS-3595 (3) in 2016 while searching for a dual-targeted p38αMAPK/JNK3 inhibitor. The two enantiomers showed similar inhibitory activity against p38αMAPK with IC50 of 0.5 μM and PDE-4 with IC50 of 0.2 μM. Moreover, MKK6-mediated phosphorylation of p38αMAPK was not inhibited at concentrations up to 10 μM. CBS-3595 reduced TNF-α release more effectively in vivo in rat lipopolysaccharide (LPS) models than p38αMAPK or PDE-4 selective inhibitors. Compared with the control group, TNF-α synthesis was inhibited by almost 80% after administration.110 It also had good pharmacokinetic properties when studied in vivo at 10 mg dose (Tm = 2.20 ± 1.30 h, Cm = 0.20 ± 0.06 μM, AUC0 − t = 0.95 ± 0.31 μM h, T1/2 = 2.7 ± 0.5 h). The researchers also found111 that treatment with CBS-3595 significantly reduced the production of the proinflammatory cytokine IL-6 and broadly increased levels of the anti-inflammatory cytokine IL-10 in rat paws. The downside, however, is that further dose increases are limited by vomiting, which is a known side effect of all PDE-4 inhibitors and thus prevents further clinical trials.
Most pyridinyl imidazole inhibitors inhibit hepatic (CYPs) enzymes. The development of ML3403 (4), a p38 inhibitor pretending to have low activity against cytochrome P450 (CYP450) enzyme, is particularly important. ML3403, originally a candidate compound in a multisubstituted pyridinyl imidazole inhibitor developed by Laufer et al.112 in 2003, was shown to inhibit TNF-α-induced strong p38αMAPK activation, and phosphorylated p38βMAPK was also inhibited by it. However, the inhibitory effects of p38γ and p38δ have not been investigated.112 The IC50 value of ML3403 for p38α was 0.38 μM, while that for TNF-α was 2.7 ± 0.3 μM, and that for IL-1β was 0.99 ± 0.46 μM.112 Similar to other inhibitors, ML3403 also inhibited TNF-α induced IL-6 secretion, and the inhibition rate was 47.8 ± 2.4% at the concentration of 1 μM. ML3403 significantly inhibited IL-8 secretion at 0.1–3 µm concentrations, but ML3403 at 10 µM did not seem to have an inhibitory effect. Pharmacokinetic evaluation of ML3403 after oral administration of 30 mg/kg in male and female Wistar rats showed rapid and high turnover of the active sulfoxide metabolite, and also demonstrated that the isoenzymes CYP1A2, CYP2C19, CYP2D6, and CYP3A4 are key enzymes in ML3403 metabolism.113 For example, in female mice, Cm reached 386 ng/mL, Tm = 1 h, AUC0 − t = 1479 ng·h/mL, while in male mice, Cm reached 115 ng/mL, AUC0 − t = 389 ng·h/mL. In another group of chronic arthritis induced by Complete Freund's adjuvant, ML3403 treatment did not show obvious hepatotoxicity.111 All these results suggest that ML3403 is a potential inhibitor of pyridine-imidazole p38α.
SB242235 (5) is a pyridyl imidazole inhibitor developed by SmithKline in 2000. The IC50 of SB242235 (5) is 19 nM for p38α, 1 μM for p38β, and almost insensitive to p38γ and p38δ, with IC50 > 10 μM.114 It inhibited p38α, p38β, JNK2β1, and c-raf, but did not inhibit ERK and JNK. And it has no direct effect on 5-lipoxygenase or cyclooxygenase-1. It competes with ATP to inhibit p38α by forming a 4-fluorophenyl-binding sac behind the site normally occupied by ATP adenine rings.115 Badger et al.116 demonstrated good inhibition of LPS-induced TNF-α production, and the use of SB242235 at 60, 30, and 10 mg/kg doses in adjuvant arthritis (AA) rats significantly inhibited foot swelling (inhibition rates were 73%, 51%, 19%, respectively). Levels of IL-6 in different inflammatory areas are sensitive markers of disease activity, and this cytokine was significantly inhibited at 60 mg/kg and not at lower doses after therapeutic administration of SB242235. Ward et al.117 studied its pharmacokinetic and showed high plasma clearance (56 ± 8.9 mL·min-1·kg−1) in rats after intravenous injection of 1.5 mg·kg−1 SB24235. T1/2 was 93.5 ± 11.3 min, Tm = 60 min, Cm = 280 ± 31 ng·mL−1.
In 1999, Wadsworth et al.118 developed RWJ67657 (6), which selectively inhibited p38α and p38β subtypes but not p38γ and p38δ subtypes, with IC50 = 1 μM for p38α. It can inhibit the production of TNF-α and IL-1β in LPS-stimulated human polymorphonuclear cells, with IC50 values of 3 and 11 nM, respectively, and block p38 signals by inhibiting the phosphorylation of p38α and downstream p38α signals. Reduced activation of p38 leads to decreased activation of downstream effectors Hsp27 and MAPAPK. It also inhibits ER signaling independently of Thr311 phosphorylation site.119 After oral administration in male and female beagles, RWJ67657 was rapidly absorbed with mean Tm values of 0.8 and 0.6 h, respectively, and disappeared with mean terminal half-lives of 3.8 and 6.1 h. The average bioavailability of the drug is about 11%. J.W. Antoon et al.120 proved that RWJ67657 effectively inhibited p38 pathway in MDA-MB-361 breast cancer cells through in vitro and in vivo experiments, and could effectively inhibit the growth of endocrine-resistant breast cancer tumors. In the presence of estrogen, RWJ67657 resulted in an approximately 3.5-fold reduction in tumor volume in the mouse xenograft model compared to tumors treated with terminal vectors. In the absence of estrogen, compound 10 reduced tumor volume by about 4.5 times after 40 days of treatment.
Ali et al.121 redeveloped a series of imidazolyl pyridine inhibitors against B-RAFV600E to inhibit p38α kinase through a drug reuse strategy in 2021, and finally developed compounds 7 and 8 as the most potent inhibitors, with IC50 of 47 nM and 45 nM for p38α, respectively. In the process of its development, considering that p38α kinase and B-RAF kinase are key components of the MAPK pathway, there is the possibility of further development. SB203580 (1), a more classical imidazolyl pyridine inhibitor, was used as the lead compound, and its imidazolyl 4-linked benzene ring was first replaced. Substitution with a hydroxyl group was found to exhibit slightly higher inhibitory activity against compounds containing 3-methoxy phenyl groups. The pyridine group was further optimized and replaced, and the propyl linker had a significant increase in inhibitory activity compared with the ethyl linker. Finally, the phenyl terminal 7 and 8 were modified, both of which showed a better inhibitory ability on p38α. In addition, the developed compound 8 also effectively inhibited LPS-induced production of proinflammatory cytokines TNF-α, IL-6, and IL-1β in RAW 264.7 macrophages with IC50 values of 78.03 nM, 17.6 µM and 82.15 nM, respectively. Compound 7 showed good activity for TNF-α production with IC50 = 98.02 nM, but did not show significant activity for the production of IL-6 and 1Il-1β. These two compounds were also developed based on the classical pyridyl imidazole structure, which further indicated that the structure played a good enlightenment role in the development of p38α inhibitors and had the possibility of further optimizing the structure.
Pyrazole derivatives
By optimizing the substitution of the lead compound SC74102 (9) on the pyrazole ring, Pfizer obtained compound 10, which significantly improved the inhibition effect on p38α and p38β. Then, by optimizing the substitution of phenyl, compound 11 was obtained, and compound 12 (SC79659) was obtained on the piperidine ring. Pyridine rings were optimized to pyrimidine rings, resulting in compound 13 (SD0006) (Figure 2).122 Compounds 12 and 13 are highly effective diarylpyrazole p38α inhibitors. The IC50 values of SD0006 for p38α and p38β are 110 and 3450 nM, while the IC50 values of SC79659 for p38α and p38β are 111 and 1790 nM, indicating that both of them have good selectivity for p38α. Inhibitor binding at ATP-binding sites can be found in the crystal structure of SC79659, while 4-chlorophenyl is tightly bound in a deeply buried lipophilic pocket, which may give SD0006 higher kinase selectivity than most ATP-competitive inhibitors. SD0006 is a highly selective p38α inhibitor that does not directly inhibit MKKs, nor does it inhibit ERK and JNK pathways. It has shown good oral anti-inflammatory effects in preclinical studies, inhibiting the expression of a variety of proinflammatory proteins and improving disease severity in animal models of arthritis, in their experiment using SD0006 to treat arthritis in rats, computed tomography was seen to significantly prevent inflammation-mediated joint and bone destruction in the 21-day treatment group compared to the control group. Burnette et al.123 reported that SD0006 almost completely blocks LPS-induced TNF α release in U937 cells in a dose-dependent manner. Similarly, it is shown in human primary monocytes and human whole blood cells to inhibit IL-1β and IL-6 similarly in U937 cells. For example, the IC50 of IL-1β and IL-6 collected from human primary monocytes is 105.9 ± 88.3 nnol·L−1 and 106.9 ± 25.3 nmol·L−1. In rats, the dose of 5 mg/kg showed good pharmacokinetic properties (Cl = 4.96 mL/min/kg, Vd = 1.74 L/kg, T1/2 = 4.5 h). In conclusion, SD0006 is a potential drug for clinical trials.

Goldstein et al.124 developed a selective p38α inhibitor RO3201195 (16) in 2005 through high-throughput screening and structure–activity relationship (SAR) studies (Figure 2), whose IC50 against p38α was about 0.7 μM. In the process of development, compound 14 was first identified by high-throughput screening. After SAR analysis, p-fluorine substitution 15 was screened out, and the IC50 of p38α was 1.1 μM, indicating that the para substitution on N-phenyl was superior to the ortho and meso substitution. Subsequently, the alternate nonionizing solubilizing functional groups were used as the main focus of the optimization strategy to obtain the final compound 16. Compound 16 also inhibited the production of rat cytokine IL-6 and TNF-α induced by LPS stimulation, which not only inhibited the production of TNF-α, but also regulated TNF-α signaling. The two hydrogen bonds in its structure are particularly important. One is based on the formation of a hydrogen bond between benzoyl oxygen and the NH backbone of methionine 109 on a skeleton called benzoyl amiprazole. And the other one is the NH2 group of aminopyrazole and the side chain alcohol of threonine 106. This is the first hydrogen bond found in p38 protein and its inhibitor. Since only about 20% of human kinases have threonine in this location, perhaps such hydrogen bonding can give it more efficient selectivity. Bagley et al.125 explored its inhibitory effect on Werner syndrome cells through experiments. In the experiment, the proliferation of dimethyl sulfone-treated fibroblasts was very slow, and only doubled in a single population during the 88-day experiment. However, the inhibitor-treated cells had a much higher rate of proliferation and controlled about 7.5 population multiplicators before growth stalled. The pharmacokinetic data were measured at 10 mg/kg in rats, Tm = 3 h, Cm = 4.48 μg/mL, T1/2 = 1.64 h, F = 61.7%, Cl = 0.32 L/h/kg, Vdss = 1.27 L/kg. In addition to the development of the inhibitor itself, Goldstein et al.124 first used the diol component as the solubilizing group of a selected kinase inhibitor in clinical studies. The data indicate that diol can be effectively used to improve the physical properties of other insoluble kinase scaffolds.
DP-802 (17), developed by Deciphera Pharmaceuticals in 2010, is a novel inhibitor that induces enhanced type II conformation when combined with unphosphorylated or diphosphorylated forms of p38α kinase. The IC50 values of DP-802 for unphosphorylated p38α and PP-p38 were 9 and 11 nM. Evaluated across a wide range of kinases, it was found to inhibit only a small number of kinases in addition to p38α and p38β, This includes the wild-type B-RAF oncogene serine/threonine kinase (IC50 = 17 nM), C-RAF oncogene serine/threonine kinase (IC50 = 11 nM) and EPH receptor B2 (IC50 = 82 nM). For the nonphosphorylated p38α-induced type II conformation, DP-802 forms a hydrogen bond with the posterior guanidine nuclear warhead of arginine 70 in the binding conformation of DP-802 and p38α (PDB:3NNW) (Figure 2). The urea part of DP-802 forms hydrogen bonds with conformal C-helical glutamate 71/lysine 53 salt bridges, tert-butyl parts occupy hydrophobic DFG sacs, 2, 3-dichlorobenzoic rings occupy type II hydrophobic sacs, and stabilize the phenylalanine 169 side chain of DFG in edge-to-facial interactions. This binding mode of DP-802 further stabilizes the p38αII conformation.126 The ability of DP-802 to inhibit the release of TNF-α stimulated by LPS in Lewis rats was also evaluated. The ED50 of DP-802 to inhibit the release of TNF-α in Lewis rats was 3.6 mg/kg 2 h after oral administration. In addition, DP-802 reduced the amount of phosphorylated p38α in HELA cells in a dose-dependent manner, suggesting that inhibitor-bound phosphorylated p38α was more readily dephosphorylated by cell phosphatases than carrier controls. DP-802 and its analogs showed strong inhibition of p38α in vivo.126
SC-409 (18), developed by Pfizer in 2005–2006, is a small molecule inhibitor of p38 with a diarylpyrazole structure. It selectively inhibits p38α and p38β isoforms with IC50 values of 0.04 and 2.30 μM, but has no significant inhibitory effect on p38γ and p38δ. SC-409 inhibits the production of key cytokines, such as TNF-α, IL-1β, receptor activator of nuclear factor-kappa B ligand (RANKL), and so on. SC-409 selectively blocks p38 signaling in pre-OCL without interfering with JNK and NF-κB pathways, and SC-409 inhibits a variety of inflammatory signaling factors including RANKL, IL-6, IL-1β, mRNA expression of tartrate-resistant acid phosphatase, matrix metalloproteases-9, -13, and COX-2. They also proved that SC-409 decreases bone destruction, joint infiltration by inflammatory cells, and the number of Ocls in streptococcal cell wall-induced arthritis in rats. In another study of cardiac remodeling in mice with myocardial infarction and heart failure, chronic in vivo administration of SC-409 improved cardiac function in patients with heart failure after myocardial infarction.
Thiazole derivatives
TAK-715 (19) is an oral p38α inhibitor for RA developed by Takeda Pharmaceutical Company in 2005, showing a good inhibitory effect (Figure 2), with IC50 = 240 nM for p38α and IC50 = 240 nM for LPS-stimulated THP-1 to release TNF-α. The inhibition rate of TNF-α was 87.6% at 10 mg/kg in LPS-induced mice, and there was no inhibitory activity against major CYPs (including CYP3A4). In its development process, Miwatashi et al.127 believe that the substitution of pyridine ring site 2 is critical to the inhibitory activity of p38 MAP kinase, and the introduction of electron-absorbing substituents or lumps enzymes is a very effective way to reduce the inhibitory activity of CYPs. The hydrogen atoms of the amino group act as hydrogen donors and bind tightly in the enzyme binding region. Several amide derivatives showed good cellular activity and pharmacokinetic characteristics, and the optimal substitution product TAK-715 was determined. In the crystal structure of p38α and TAK-715 (PDB: 3ZSG), the pyridine N atom of the inhibitor forms a hydrogen bond with the main chain NH of Met109. The amide NH of TAK-715 forms a hydrogen bond with the main chain carbonyl group of Met109. The N atom of the thiazole nucleus forms a direct hydrogen bond with Lys53, the active site, which also interacts with Glu71. The thiazole nucleus is deposited on the phenyl side chain of Phe169, and the ethyl group of thiazole is within the van der Waals distance of Tyr35. To verify the inhibitory effect of compound 19 on LPS-induced TNF-α production in mice and its pharmacokinetic parameters, Cm = 0.19 ± 0.05 μg/mL and AUC0–24 h = 1.16 ± 0.16 µg·h/mL at 10 mg/kg dosage. There has been a Phase II national clinical trial (NCT) (NCT00760864) on RA conducted in 2004–2005.
Hynes et al.128 developed BMS-640994(20) inhibitor in 2008, whose IC50 for p38α and TNF-α was 3.5 and 2.9 nM. For lymphocyte-specific protein tyrosine kinase (Lck), the IC50 was 22,000 nM. In the development process, BMS-640994 is based on the main structure of pyrrole [2, 1-F][1,2,4] triazine to explore, when 2-methyl-5-formamide aniline replaces 2-chloro-6-methylaniline, a >3-fold increase in the titer of p38α enzyme and human peripheral blood mononuclear cells (PBMC) was observed. When C2 substituents contained branched aliphatic groups, a trend of increased cell potency was observed, resulting in compound 20. In the x-ray diffraction structure of and p38α (PDB: 3BX5), the thiazole ring nitrogen forms a hydrogen bond with the main chain NH of Met109. Additional hydrogen bond interactions are obtained between the Met109 carbonyl group and C2-amine NH, and the C5 benzamide carbonyl oxygen can form hydrogen bonds with the main chain NH of Asp168, while Glu71's locus is far away from this region.128 In addition to blocking TNF-α, it also inhibited the production of IL-1β in PBMCs in LPs-induced mice with an IC50 of 99 nM.128 In addition, LPs-induced TNF-α and IL-1β were inhibited in whole human blood with IC50 values of 54 and 600 nM. When screened against a group of kinases, it was shown to be highly selective against p38α, and only p38α and p38β were effectively inhibited among the four p38 isoforms. The pharmacokinetic data were measured at 1 mg/kg in rats, T1/2 = 1.8 h, Cl = 14.1 mL/min/kg, Vss = 1.8 L/kg, Cm = 8.7 ± 2.2 μM, AUC0–24 h = 25 ± 2.3 mM·h. Although it was developed in 2008, no clinical trials have been conducted on it, possibly because of its potential toxicity.
Schehr et al.129 developed a photoswitch kinase inhibitor 22 of p38α in 2019 by linking an existing, well-characterized inhibitor scaffold to an arylazo and diazoxide moiety. Photoswitch molecules, which undergo conformational or configuration changes triggered by light, have shown great potential in versatile applications. Photopharmacology is to control the activity of molecules by changing their geometry by light. In the process of its development, the lead compound 21 was used as the backbone (p38α IC50 = 10 nM). Through preliminary docking, it was also analyzed that the moiety was located in a narrow channel composed of multiple amino acid residues, so it could accommodate the azaryl moiety of the E configuration but not the azaryl moiety of the Z configuration, thus converting into different photoresponsive biological activities. Finally, the 2-imidazole substituted by methyl sulfide in the existing molecule was considered to be the most suitable position for connecting the switchable azo unit, and its activity was significantly improved by replacing it with an azo unit-linked benzene ring. Then, the imidazole skeleton was replaced by a thiazole, and a new type of photoswitch kinase inhibitor was developed. Both the E and Z configurations (435 nm irradiation) of the compound showed good p38α inhibitory activity with IC50 of 29.1 ± 8.62 nM and 2.36 ± 0.61 nM, respectively. Notably, the Z isomer was more potent after photoisomerization. In the cocrystalline structure, it can be seen that the compound dominated by the E configuration, Lys53 directly forms hydrogen bonds with the N on the thiazole ring, while Met109 forms hydrogen bonds with the N on the pyridine ring and the N on the amide bond, respectively. For this newly developed photoswitch kinase inhibitor, its uniqueness and novelty deserve more long-term attention.
Bisamide derivatives
Shaun R. Selness et al. discovered a series of novel p38α inhibitors in 2011, such as PH-797804 (25) (Figure 2). In the process of its development, the 2, 5-bis substitution of compound 23 was more favorable to its inhibitory activity than the 2, 3-and 2, 4-bis substitution. Furthermore, through the optimization of N-aryl ring, the very important 2-site substitution was focused on. Thus, PH-797804 (25), a 2.3 nM IC50 inhibitor of p38α, was obtained. In the study of N-aromatic ring 2 substitution, fluorine was introduced to produce compound 26, which was more tolerant, but its activity (IC50 = 8.5 nM for p38α) was slightly lower than that of PH797804. Compound 27 was obtained by introducing chlorine, whose IC50 to p38α was 3.0 nM. It was also seen that when polar functional groups were introduced into the second site of the N-aryl ring, such as compound 28, a significant effect on potency was observed, with an IC50 of 325 nM. This loss of potency was attributed to the negative interaction of the hydrophilic 2-substituted group with the lipophilic environment formed by the G-ring and p38α extended hinge region. According to SAR analysis, small lipophile groups isomorphic to methyl groups are basically equipotential, and replacement with larger groups or more polar groups will result in loss of p38 affinity.130 The eutectic structure of compound 25 with p38α (PDB: 1CM8) suggests that PH-797804 achieves its selectivity not by inducing inactive kinase conformation, but by binding unique interactions with the hinge and the unique hydrophobic pocket of p38α kinase. Due to the spatial volume of the pyridinone carbonyl group and the 6,6′-methyl substitution of PH-797804, the torsion-rotation around the bond connecting pyridinone and n-phenyl ring is blocked to produce a discrete conformation space of the N-phenylpyridinone group, resulting in two atropine isomers that do not convert to each other under environmental conditions.131 The binding of the pyridone portion of PH-797804 overlaps with the adenine portion of ATP. In addition, the inhibitor also occupies a unique aryl pocket that binds differently to the phosphoric acid of ATP. The didentate hydrogen bonds to Met109 and Gly110 require the peptide bond to flip into a conformation suitable only for glycine residues.131 Compound 25 continues to be under clinical investigation for its excellent selectivity and high efficiency as a potential p38α inhibitor for the treatment of inflammatory-mediated diseases. For example, the phase II experiment on RA (NCT00620685) was conducted in 2008, the phase II experiment on Osteoarthritis (NCT01102660) was conducted in 2010–2011, and the phase II experiment on COPD (NCT01321463) was conducted in 2011–2012.
Losmapimod (29) inhibitor, developed by GlaxoSmithKline, Inc., is a dual p38α/β ATP-competitive inhibitor with pka of 8.1 in p38α and 7.6 in p38β, which effectively inhibits TNF-α in whole blood. As the most widely effective p38 inhibitor in clinic, it has been developed to treat RA, and is also in clinical evaluation for indications, including COPD and heart disease.132 In COPD, Losmapimod reduces acute phase circulating proteins, such as plasma fibrinogen, and has a tendency to decrease other markers of systemic inflammation, such as IL-6, IL-8, and C-reactive protein (CRP).133 P38MAPK is inhibited at the cellular level by reducing the phosphorylation level of heat shock proteins. In cardiovascular diseases, Losmapimod reduced atherosclerotic arterial inflammation,134 moderated FDG-PET/CT-associated vascular inflammation in active inflammatory segments, and significantly reduced the intake of fluorodeoxyglucose (FDG) biomarkers for circulation inflammation and FDG in visceral adipose tissue. Improvement of endothelial function in hypercholesterolemia. A series of clinical trials were conducted, such as Phase II for Focal segmental glomerulosclerosis (NCT02000440) from 2014 to 2016, and Phase II for COPD (NCT02299375) from 2014 to 2016. And the Phase II trial of Facioscapulohumeral muscular dystrophy (NCT04003974) from 2019 to 2021. In addition, the Food and Drug Administration (FDA) has granted Orphan drug status to Losmapimod for the treatment of facial shoulder brachial muscular dystrophy.
VX-702 (30), a pyridine p38α inhibitor developed by Vertex Pharmaceuticals, is a second-generation p38α inhibitor with an IC50 range of 4–20 nM. VX-702 can inhibit the production of IL-6, IL-1β and TNF-α stimulated by LPS, with IC50 values of 59, 122 and 99 ng·mL−1.135 VX-702 was developed as a treatment for RA, in clinical trials, decreased levels of CRP, soluble tumor necrosis factor receptor p55, and serum amyloid A were observed at Week 1, but these levels quickly returned to baseline by Week 4 with a higher incidence of severe infection.136 Another factor that may affect the magnitude of response is the lack of CNS penetration in VX-702. The aggregate data suggest that VX-702 inhibition of p38MAPK may not provide meaningful and sustained inhibition of RA. However, there are still some clinical trials, including the RA Phase II trial conducted in 2005–2006 (NCT00205478), and the RA Phase II trial conducted in 2006–2007 (NCT00395577).
UM-164 (31), developed by Giliani et al. is a potent dual-target Src/p38 inhibitor that inhibits both p38α and p38β isoforms with a Kd of 2.2 nM for p38α, 5.5 nM for p38β, and 2.7 nM for Src. It is the only kinase inhibitor that can effectively inhibit c-Src, p38α, and p38β, but not most kinase groups, and is a promising lead compound for the development of the first targeted therapy strategy for triple-negative breast cancer (TNBC). Gilani et al.137 found that UM-164 forced its target kinase into a specific inactive conformation (DFG-out), demonstrating that UM-164 binding to c-Src in its inactive conformation altered the localization of c-Src in TNBC cells. These effects combine to produce a potent anti-TNBC activity of UM-164. In the TNBC xenograft model, UM-164 resulted in a significant reduction in tumor growth compared with the control group.
Other monocyclic inhibitors
Nirav G. Shah et al.,138 in their 2017 study of selective inhibitors of p38α and p38β, used computer-aided drug design to target the glutamate-aspartate (ED) substrate docking pocket of p38α rather than the catalytic site. UM101 (32) was screened, and differential scanning fluorimetry and STD-NMR showed that UM101 specifically bound to p38α (Table 1), while compound 31 had only weak inhibitory activity on p38β, which may be due to the interaction between the protons in the two aromatic rings of UM101 and p38α, while UM101 and p38β have almost no aromatic interaction. Shah et al. abolished the binding of UM101 to p38α by site-directed mutagenesis of four amino acids in the binding pocket, suggesting that they are the key amino acids in the binding process. UM101 has been shown to stabilize the endothelial barrier function of human pulmonary microvascular endothelial cells. Partially blocked the phosphorylation of p38 substrate MK2 and signal transducer and activator of transcription (STAT-1) in HeLa cells stimulated by TNF-α, and inhibited the expression of proinflammatory genes in THP1 cells induced by LPS. It is well tolerated in relieving experimental acute lung injury. PathwayNet analysis showed that UM101 inhibited the expression of transcription factors (STAT-1, c-Fos (c-fos proto-oncogene), c-Jun, NF-κB, p53, PPARg and Sp1). However, it did not inhibit other transcription factors (ATF1, ATF2, ETS transcription factor ELK1, c/EBPb, human upstream transcription factor 1, small mothers against decapentaplegic 3, Forkhead box transcription factor O1.138 At present, there are few studies on UM101, but this does not affect the inhibitor potential of the developed UM101.
PNU-120596 (33) is a classical positive allosteric modulator of α7 nicotinic acetylcholine receptors (α7 nAChR, α7 nicotinic acetylcholine receptors). Uwada et al.139 demonstrated that PNU-120596 also inhibited p38MAPK phosphorylation, including that PNU-120596 pretreatment inhibited oxidative or osmotic stress-induced p38MAPK phosphorylation in 293A cells and also inhibited TNF-α-induced p38MAPK phosphorylation. The inhibition of p38MAPK phosphorylation was also confirmed to be independent of α7 nAChR, which directly inhibited p38α with an IC50 value of 3.3 μM and was insensitive to the other three subtypes. PNU-120596 also inhibited LPS-induced phosphorylation of p38MAPK and LPS-induced expression of TNF-α, IL-6, and COX-2 in BV-2 cells. Structurally, the urea-NH group and C=O group of PNU-120596 form hydrogen bonds with the p38αMAPK on the Glu71 side chain and Asp168 backbone. Therefore, the diarylurea scaffold of PNU-120596 may play a key role in binding to p38MAPK.139 The rapid binding kinetics of PNU-120596 indicated that p38MAPK binding of PNU-120596 was more likely to be DFG-in binding. In the study of improving neuroinflammation and movement in rats with PD,140 PNU-120596 inhibited striatal neuroinflammation, which was manifested as the inhibition of JAK2/NF-κB/GSk3β expression, and the expression level of TNF-α protein in nigrostriatal tissue was decreased, reflecting the anti-inflammatory ability of this substance. However, at high concentrations, PNU-120596 showed a relatively low inhibitory effect on p38MAPK.
L.k. Petersen et al.141 developed a group of triamide-type selective p38MAPK inhibitors VPC00628 (34), VPC00630 (35), VPC00257 (36) for p38α IC50 of 7 ± 0.9, 44 ± 4.4, 46 ± 2.5 nM. VPC00628, which is the most effective, is only selective to p38α and p38β, but not to other isoforms. In the binding conformation analysis of VPC00628 and p38α (PDB:5LAR), it was found to bind in a typical Type II (“DFG-out”) binding mode, and more prominently its induced folding conformation of the P-ring. Although VPC00628 lacks key features of kinase inhibitors, such as the typical hinged binding motif, it exhibits good shape complementarity structurally, where several polar interactions are particularly important, such as the formation of hydrogen bonds between its pyrazole nitrogen and the amide backbone of Met109. The amides adjacent to pyrazole interact with the carbonyl main chain of Thr106gatekeeper and P-ring Tyr35 directly and through water molecules. Cyclohexane decoration deep into the “DFG-out” conformation formed by the hydrophobic pocket; Carboxylamide at the end of the inhibitor forms hydrogen bonds with the protein main chain (Phe169 and Gly170), and so on.141
In 2020, Rohm et al.142 improved the selectivity by modifying VPC00628, an excellent p38α selective inhibitor, and created a more effective and selective p38α/β inhibitor compound 37 with IC50 of 14.0 and 16.8 nM for p38α and p38β, respectively. It is also an alternative type II chemical probe targeting p38α/β. During development, the researchers thought that the DFG back pocket binding part of VPC-00628 could be optimized by further extending it into the αC-out pocket. Compared with its lead compound VPC-00628, the NH2 residue at the end of the lead compound was replaced to extend the amide bond site to better extend the backward compartment, and both linear and branched fatty residues were introduced. After comparing various compounds with different types of amide residues and branched chain residues, compound 37 was created, which also had better thermal stability, ΔTm = 16.4 ± 0.2°C, and the inhibitory ability of p38α was also more than doubled. When selected for screening 468 kinases and mutants, the compound showed good selectivity, only seven were detected off-target, while its Kd for p38α/β was 6.3/20 nM. In addition, the compound showed a dose-dependent inhibition on the activation and phosphorylation of p38 and the phosphorylation of its downstream substrate Hsp27 in HCT-15 cells, while its inhibitory effect on TNF-α release was significantly stronger than that of compound SB-203580. The compound was also tested for metabolic stability in human liver microsomes and showed good stability, indicating its excellent optimization potential.
In the process of discovering BTK and LCK inhibitors in 2017, Wang et al.143 used p38α as a model system for structure-based virtual screening and developed a potential triazole p38α inhibitor 38 with an IC50 of 14.445 μM against p38α. The IC50 for BTK and LCK was 1.091 and 0.531 μM, respectively. Wang et al. performed a structure-based, layered virtual screening of the ATP-binding sites of p38α from (1) hydrogen bonds to the hinge de-backbone atoms including the carbonyl group of His107 and the amide group of Met109, (2) residues with good contacts around the hydrophobic pocket including Val38, Leu75, and (3) residues with good contacts around the hydrophobic pocket. Two kinase-specific filters, Ile84 and Leu104, were used to screen, among which compound 38, in its cocrystalline structure with p38α (5XYX), formed four hydrogen bonds at the hinge region, Met109, His107, and Thr106, which formed one, two, and most of the available inhibitors. The few three hydrogen bonds are particularly different. This inhibitor belongs to the inhibitor obtained by virtual screening, and further optimization potential is needed to improve its selectivity and inhibitory activity.
4.1.2 Bicyclic p38α and p38β inhibitors
BIRB-796 (42), developed by Boehringer-Ingelheim, Germany, is the earliest highly selective inhibitor of p38α entered the phase II clinical trial (Figure 3). Its IC50 against p38α and p38β were 38 and 65 nM. BIRB-796 inhibited the production of TNF-α under LPS stimulation, and the IC50 value was 21 nM. In the process of its development, compound 39 was used as the substrate to optimize the imidazole group replacement, and the addition of isobutyl group made compound 40 have a higher Tm than compound 39. According to studies, imidazoline-2 substitution is particularly important for its inhibitory effect. After screening, methyl benzene was selected to replace compound 41, and finally ethoxy morpholine was used to replace H on naphthalene to get the final product BIRB-796. For its structure, the naphthalene group can be used as a kinase-specific pocket, and the tert-butyl imidazole part can be used as a phenylalanine pocket. The x-ray structure of p38αMAPK and BIRB-796 (PDB:1KV2) showed that Asp168 and Leu167 had hydrogen bonding on the carbonyl group of urea, Glu71 had hydrogen bonding with the NH group of urea, and Met109 had hydrogen bonding with the O of morpholine group. Both Phe169 and Lys53 interact with naphthalene group π–π, and the conserved residue Asp168-Phe169-Gly170 (DFG) moves to a new location during binding. In the new conformation (DFG-out), the binding of ATP to p38α MAP kinase is inhibited. However, in later clinical trials, the researchers stopped the clinical studies related to BIRB-796 due to toxic side effects such as elevated liver enzymes. In view of the good activity of BIRB-796, Arai et al.145 carried out a series of modifications on the original BIRB-796 in 2012, replacing the naphthalene group of BIRB-796 with benzyl group, and confirmed that compound 43 binds to the allosteric site of p38αMAP kinase similar to compound 42. The IC50 of compound 43 for p38α reached 12 nM. And benzyl rings occupy kinase-specific pockets. Therefore, compounds 44 and 45 are further modified to obtain the IC50 of 4 and 5 nM for p38α. Ryoo et al.146 experimentally demonstrated that BIRB-796 can inhibit the secretion of cytokines by THP-1 cells induced by LPS, and inhibit the transcription of cytokines induced by LPS and the phosphorylation of p38αMAPK in THP-1 cells. The therapeutic effect was evaluated by measuring the changes of alanine aminotransferase (ALT) and aspartate aminotransferase (AST) in blood. After 5 days of oral administration, ALT content was 24 ± 4 U/L and AST content was 45 ± 11 U/L. Later, in a series of studies, it was confirmed that BIRB-796 could inhibit not only p38α, but all subtypes of p38, but only p38α and p38β at low concentrations. In addition, a series of clinical trials have been conducted under better efficacy, such as the phase II trial on Psoriasis (NCT02209753) conducted as early as 2001–2002, and the phase II trial on RA (NCT02209779) conducted in 2001–2002. All with good results.

The IC50 of VX-745 (49) was 9 nM for p38α, 45 nM for IL-1β, and 51 nM for TNF-α. It blocks the release of IL-1β and TNF-α in whole blood with IC50 values of 150 and 180 nM. VX-745 is highly selective, with p38α being 20 times more selective than p38β (Ki = 220 nM), and has no significant inhibitory effect on other MAP kinases (except MKK6). John P. Duffy et al.147 performed SAR analysis on compound 46. As the lead compound, compound 46 showed IC50 for p38α > 20 μM, but IC50 for IL-1β and TNF-α was not accurately detected. The benzene ring was optimized by substituent group, and it was determined that the chlorinated product 47 at position 2 had an obvious decrease in IC50. Further substituent group optimization was carried out to determine that the 2, 6 dichlorobenzene ring substituted compound 48 had a stable interaction with p38α, and the best substitution effect of 2, 4 difluoride on another benzene ring was determined by analysis, namely compound VX-745. Compound 49 occupies a hydrophobic pocket with each chlorine atom and is in contact with residues Val30, Leu108, Ala157, and Leu167. The benzene ring itself interacts favorably with the main chain residue atoms Gly110, Ala111, and Asp112. 2, 4-difluorophenyl rings occupy hydrophobic pockets at gatekeeperThr106 residues and form extensive van der Waals contacts.147
PF-03715455 (50), an inhibitor developed by Pfizer, is selective to p38α and p38β, with IC50 of 0.88 and 23 nM148 (Figure 4). It had long-term effects in vitro (t½ > 80 h) and in vivo, and effectively inhibited LPS-induced TNF-α production in human blood (IC50 = 1.7 nM). Compound 50 binds to p38α quickly, dissociates slowly and has a long half-life. From a structural point of view, it may be due to the larger group at the 2-site of triazopyridine (2-hydroxyethyl thiophenyl and isopropyl) leading to increased affinity. And the structure of 50 has been optimized with a “design inhalation” approach designed to minimize systemic exposure and improve therapeutic indices. PF-03715455 is a very effective and selective inhibitor in the treatment of COPD, which can inhibit the release of proinflammatory cytokines associated with COPD. The kinetic parameters of p38α were t1/2 = 80 h, KD = 0.001 nM. At the same time, its pharmacokinetic characteristics in vitro and in vivo indicate that oral bioavailability of this compound in humans is very low at any swallowed dose and has a high intrinsic clearance, so that any drug absorbed by the lungs is rapidly metabolized into relatively inactive metabolites. PF-03715455 was tested in a phase I trial on COPD as early as 2011 (NCT01314885).

In 2004, Palau Pharmaceuticals released a novel pyrazolypyridine inhibitor UR-13756 (51). UR-13756 showed higher kinase selectivity through a kinase set of 115 kinases compared to BIRB-796 (42) and SB203580 (1). Especially regarding JNKs and c-Raf1.149 UR-13756 has a higher selectivity for p38α than p38β, and its IC50 for p38α is about 80 nM. For UR-13756 to inhibit the production of TNF-α by LPS-induced peripheral blood mononuclear cells, the IC50 in this system was comparable to the EC50 at 60 nM, and in the same study, the EC50 of SB203580 was 280 nM. In addition, 1.0 µM UR-13756 was 100% effective in inhibiting p38α after pretreatment for 24 h.149 The clinical efficacy and safety of UR-13756 was described in the first reported large-scale randomized clinical trial of a p38 inhibitor with good bioavailability, favorable pharmacokinetic properties, and low toxicity.150, 151
Peifer et al.152 developed a selective p38α inhibitor, CP41 (52), in 2007. The IC50 for p38α was 1.8 μM, and two isoxazole isomers were developed simultaneously. The IC50 for p38α of compound 53 was 0.45 μM, the IC50 for JNK3 was 0.54 μM, the IC50 for JNK2 was 1.28 μM, and the IC50 for p38α of compound 54 was 2.2 μM. IC50 of JNK3 = 3.5 μM. CP41 only inhibited p38α subtype of MAPK, while 53 and 54 separately blocked JNK2/3. In addition to MAPK, compound 53 also significantly inhibits Protein Kinase A (PKA), Protein Kinase D1 (60% ± 1), Lck (42% ± 0), and casein kinase 1 enzyme δ (6% ± 2). However, CP41 did not inhibit TNF-α-induced IL-6 protein secretion nor significantly inhibited TNF-α-induced IL-8 secretion. In its binding conformation model (PDB: 1A9U, PDB: 1PMN), the o-pyridine-4-f-phenyl system in CP41 favors the inhibition of p38α. Compared with Thr106 in p38α, the action of the more spacious Met146 in JNK3 determines the selectivity of the six-membered core compound CP41 for p38α. But not for the five-membered core compounds 53 and 54.152 Although no clinical trials have been conducted, CP41 is a good lead product for the development of selective inhibitors of p38α.
SCIO-323 (55) is a small molecule of indolyl benzylpiperazine formamide that acts as an ATP-competitive inhibitor of p38MAPK kinase. The IC50 of p38α and p38β was 3, 180 nM, and p38γ and p38δ were not significantly inhibited. S.B. Goodman et al.153 studied the efficacy of SCIO-323 in reducing the established inflammatory response to periprosthesis osteolysis caused by nonsurgical treatment of wear particles and demonstrated that oral SCIO-323 helps reduce inflammatory response to wear particles. And to some extent reverse the adverse effects of phagocytic polyethylene particles on bone growth in our rabbit model. However, continuous administration of SCIO-323 may interfere with the production of key cytokines and growth factors important for the initial stage of bone formation, adversely affecting the proliferation, differentiation, and maturation of mesenchymal tissues. But with the deepening of research in recent years, SCIO-323 is gradually found to be ineffective in vivo.
Dilmapimod (SB-681323) (56) has been identified as a potent, selective, and reversible inhibitor of p38α MAPK, and has been shown to inhibit the production of several cytokines such as IL-6, IL-8, and so on. In the treatment of COPD, Dave Singh et al.154 proved that Dilmapimod could effectively inhibit p38MAPK signal by reducing the expression of phosphorylated heat shock protein 27 (pHSP27) induced by sorbitol, which in turn inhibits the production of TNF-α. In clinical trials for severe trauma patients at risk of acute respiratory distress syndrome, Jason D. Christie et al.155 demonstrated that intravenous Dilmapimod was generally well tolerated in severely injured subjects at risk of ARDS, and at the dose levels tested, there is no evidence of hepatotoxicity after treatment of trauma patients, whereas other partial p38MAPK inhibitors cause an increase in liver transaminase. At the same time, IL-6, IL-8, CRP, and soluble tumor necrosis factor receptor-1 levels have been reduced in the trial, and these changes are most obvious in the high-dose continuous infusion scheme. Dilmapimod has conducted several clinical trials, such as the Phase I trial of COPD (NCT00380133) (NCT00564746) in 2005–2006 and 2007.
Pamapimod (RO-4402257) (57) was a selective and potent inhibitor of p38α and p38β with IC50 values of 0.014 ± 0.002 μM and 0.48 ± 0.04 μM, but had no significant effect on p38γ and p38δ. It is a prototypical ATP-competitive kinase inhibitor that binds to the hinge region and interacts with the anterior and posterior capsules of the ATP-binding site of p38. Pamapimod also inhibited LPS-stimulated TNF-α, IL-1β produced in whole blood and spontaneous TNF-α produced in RA patients. It was also shown that Pamapimod inhibits phosphorylation of HSP27, a MAPKAPK-2 substrate, which directs p38 signaling through MAPKAPK-2. A Pamapimod binding assay for 350 other protein kinases156 showed that the molecule is highly selective and binds only to another MAPK family member, JNK, with an affinity comparable to that of p38. However, despite the apparent affinity for JNK, the evaluation of JNK inhibition based on c-Jun phosphorylation in both cell types indicated that Pamapimod was ineffective against JNK in cells. Thus, Pamapimod in vivo is expected to have little direct effect on JNK activity. In the process of clinical trials on the treatment of RA, it was found that compared with methotrexate, the disease response rate of patients with active RA treated with single drug Pamapimod was reduced and the incidence of adverse events was increased, which was obviously not suitable as a substitute for methotrexate. Therefore, it was considered as a possible supplement to methotrexate treatment.157 In conclusion, Pamapimod is a highly selective inhibitor of p38α/β and the anti-inflammatory activity of Pamapimod both in cells and in vivo suggests its potential to alleviate inflammation.
The inhibitor Talmapimod (SCIO-469) (58) developed by SCIOS is a potent p38α inhibitor that is more than 1000 times more selective than ERK2, JNK1, and Lck. The distinctive feature of indoleamides is that they can achieve high selectivity to p38α and p38β with IC50 of 14 nM for p38α and 480 nM for p38β. Talmapimod also moderately inhibited LPS-induced TNF-α production in human whole blood with an IC50 of 300 nM. The binding mode of Talmapimod and p38 involves the C5 carbonyl group interacting with p38 Met109 via hydrogen bonding.158 The lipophilic benzylpiperazine is thought to occupy the selective pocket of the kinase. The o-substituent on the indole ring may help to achieve high p38α selectivity. The adjacent substituent occupies a hydrophobic pocket near the Ala40 residue of p38α, whereas p38β has a larger, more hydrophilic serine residue in a similar position. In rats with experimental arthritis, Talmapimod reduced signs and symptoms of the disease and slowed the progression of the disease. However, in clinical trials, the use of Talmapimod in patients with RA produced only a transient benefit, and no long-term effective benefit. In more recent studies, such as the phase II trial of RA (NCT00043732) in 2003, from 2004 to 2006, the phase Ⅱ trial (NCT00095680) and other studies on multiple myeloma (MM) gradually found that Talmapimod had poor efficacy in vivo.159
SX-011 (59) has 10-fold p38α/β selectivity, whereas the azindole derivative of SX-011 has 249-fold p38α/β selectivity, making it one of the most selective p38α inhibitors reported.
Ralimetinib (LY2228820) (60) is a trisubstituted imidazole derivative that is a potent and selective ATP-competitive inhibitor of the p38α and β isoforms (IC50 = 5.3 ± 1.6 and 3.2 ± 0.3 nM) (Figure 4). In cell-based assays, Ralimetinib effectively and selectively inhibited p38α MAPK substrate phosphorylation (p-Thr334-MK2) without affecting phosphorylation of p38α MAPK, JNK, ERK1/2, c-Jun, ATF2, or cMyc. It can also effectively reduce LPS-stimulated cytokines secreted by macrophages and A549 NSCLC cells.160 Ishitsuka et al.161 explored the therapeutic potential of Ralimetinib for MM, it was confirmed that it inhibited the secretion of IL-6 from bone marrow stromal cells and bone marrow mononuclear cells (BMMNC) of patients with MM in remission. LY2228820 also inhibited macrophage inflammatory protein-1a secretion in MM cells and BMMNC from patients and in normal CD14-positive osteoclast precursor cells. In addition, LY2228820 significantly inhibited macrophage colony-stimulating factor and NF-κB ligand-soluble receptor activator-induced osteoclastogenesis in CD14-positive cells in vitro.162
SKF-86002 (61) is the lead product of SB203580 (1), the IC50 of SKF-86002 is 0.26, 0.50, 0.40 μM for p38α, IL-1β, and TNF-α. In its bicycloimidazole compounds, the correct regional chemistry on the core heterocyclic ring is essential for effective binding of p38, confirming that only on the imidazole ring close to the pyridine ring is nitrogen tolerated as a substitutant without loss of nitrogen activity. The inhibition of IL-1β by SKF86002 showed that SKF86002 could only reduce eight peptides produced by monocytes. Inhibition of IL-1β production was achieved by influencing two independent steps in the biogenesis of this cytokine. One is that SKF86002 reduces the synthesis of proIL-1β, and the second effect of SKF86002 is to inhibit the release of IL-1β by affecting high concentrations of LPS.163 In the treatment of streptococcus M1 protein-induced neutrophil activation and lung injury,164 M1 protein activation increased the phosphorylation and activity of p38MAPK in the lung, while SKF 86002 reduced M1-induced neutrophil infiltration, pulmonary edema, and CXC chemokine formation by inhibiting p38MAPK activity, and the upregulation of neutrophils by Mac-1. Recently, Masliah's team165 used SKF-86002 to treat DLB/PD in vitro cell and mouse models and found that it promoted the redistribution of p38γ to synapses and reduced α-synuclein accumulation, neuron loss, and motor deficits in the mouse brain.
Kaieda et al.166 in 2019 used a structure-based design strategy to identify a series of p38MAPK inhibitors through high-throughput screening, including the discovery and design of a derivative of imidazopyridinone 65 that is a potent inhibitor of p38MAPK (p38α IC50 = 9.6 nM) and a derivative of imidazopyridinone that is a potent inhibitor of p38MAPK. It also showed excellent inhibitory effect on TNF-α production (THP-1 IC50 = 46 nM) and TNF-α induced IL-8 production (hWB IC50 = 15 nM) in human monocytic leukemia cells. During its development, the lead compound 62 was first selected by high-throughput screening with an IC50 of 140 nM for p38α. On the basis of the cocrystal structure analysis of p38α, which is thought to interact with Met109 and Gly110 in a bidentate manner, several scaffolds were first explored, including hydroxyindole, aza-indole, benzimidazolone and imidazopyridin-2-one. Compound 63 with imidazopyridin-2-one as the basic skeleton was found to be relatively different from other scaffolds. It has a better inhibition ability (IC50 = 390 nM). When compound 63 was further optimized, it was found that the substitution group on the terminal benzene ring was important for improving the inhibitory activity, so a more excellent compound 64 with IC50 = 63 nM was obtained by 2, 4-difluoride substitution on benzene. Finally, the most excellent compound 65 with IC50 = 9.6 nM was obtained by optimizing the substituent of imidazole. In the cocrystalline structure of compound 65 and p38α, aniline hydrogen binds Lys53 and Asp168 via water-mediated hydrogen bonds, and 2, 4-difluorophenyl enters the shallow inner hydrophobic pocket of p38MAPK composed of Thr106 and Leu104. Pharmacokinetic study in rats showed Cm = 55.8 ng/mL, Tm = 2.7 h, AUC = 223.2 ng·h/mL. After comprehensive evaluation of its inhibitory effect, solubility, and pharmacokinetics, this compound was considered to be the best potential lead compound in this series of imidazo[4, 5-B]pyridin-2-one-based p38 MAP kinase inhibitors.
The IC50 of AZD6703 (66) was 16 nM for p38α and 0.027 μM for HWB (Figure 5). AZD6703 was originally developed as a bisamide inhibitor. The earliest compound 67 had related inhibitory activities, but its water solubility was poor and plasma protein binding was quite high. Previous studies have shown that its terminal heteraryl methoxy can be removed without significantly reducing potency and replaced by side chains/rings containing alkaline amines, thereby improving solubility and plasma protein binding.167 D.S. Bowen et al.167 obtained a novel inhibitor AZD6703 by sequentially converting two aniline embedding functions in the diamide series into quinazolinone and N-cyclopropyl amide. Physical and PK properties are improved by introducing weakly basic arylpiperazine groups and reducing molecular weight, and activity in human whole blood is improved by reducing plasma protein binding. Interaction of piperazine basic nitrogen with Val30 amido-carbonyl group in eutectic structure of AZD6703 and p38α (PDB: 4AA5). The structural study showed that the inhibitor changed from “DFG out” mode to “DFG in” mode, and was of I½ type. In 2020, Raubo et al.168 believed that the alternative groups of N-methylpiperazine in AZD6703 should also be easy to study, and compound 68 of 3, 5-dibromo-2(1H)-pyrazinone was selected as a new inhibitor framework for study, resulting in a series of new inhibitor types.

The IC50 of R1487 (69) is 10 ± 4 nM for p38α and 200 ± 87 nM for HWB. For kinase selectivity, Kd for p38α is 0.2 nM and Kd for p38β is 29 nM, which does not bind to p38γ and p38δ. In the presence of compound 69, detailed Kd assays of kinases inhibited in a single spot screening confirmed that only 10 of the >300 kinases tested were submicromole inhibited. The lead compound of R1487 is compound 70, was originally designed as an inhibitor of lymphocyte-specific protein tyrosine kinase (Lck), a member of the src kinase family, with Kd = 0.2 nM for Lck and Kd = 10 nM for p38α. In SAR analysis, incorporation of saturated alkyl amines at the 2-site increased the three-dimensional volume of the molecule and led to adverse spatial interactions between inhibitors and kinases containing glycine insertions in the lower lip region. To further enhance the selectivity of kinase, additional template modification was carried out to expand the volume of the posterior hydrophobic capsule of p38α by inserting oxygen atoms between the core scaffold and the aromatic ring.169
BMS-582949(PS-540446) (71) was developed by Liu et al.170 in 2010. The IC50 was 13 nM for p38α and 50 nM for hPBMC TNF-α. Its lead compounds pyrrole [2, 1 f] [1, 2, 4] triazine derivatives 72 and 73 are potent p38αMAPK kinase inhibitors, however, compounds 72 and 73 showed high clearance in vivo. In the course of its studies, the N-methoxy benzamide functional groups in 72 and 73 could be partially replaced by urea, carbamates, and reverse amides without methoxy amino groups, resulting in a number of highly potent p38αMAPK kinase inhibitors. The cocrystal structure of BMS-582949 and p38α (PDB: 3MVM) shows that the binding mode of N-cyclopropyl benzamide is similar to that of 72. The benzamide moiety provides two key hydrogen bond interactions with p38α: one with the carbonyl oxygen of Asp168 and the NH interaction with Glu71. Another important hydrogen bond interaction occurs between the 6-carboxamide oxygen and Met109 in the hinge region. Hydrophobic interactions include the angular methaniline moiety located in the hydrophobic pocket, and the n-propyl group interacting with the typical hydrophobic point on the outer edge of the hinge region.170 Evaluated in a sham rat model of AA, BMS-582949 showed dose-dependent reduction in foot swelling, with efficacy seen at doses of 10 and 100 mg/kg. In addition, in the human hepatotoxicity assay, the IC50 of all its subtypes were detected greater than 138 μM, demonstrating no cytotoxicity and suggesting a reduced likelihood of clinical hepatotoxicity. For clinical trials, a phase I trial for RA (NCT00162292) was conducted from 2005 to 2007, and a phase II trial for Psoriasis (NCT00399906) was conducted from 2007 to 2009.
4.1.3 Tricyclic p38α and p38β inhibitors
Skepinone-L (74) is a dibenzocycloheptanone compound developed by Koeberle et al.171 in 2011 (Figure 6), which shows excellent effect selectivity in the inhibition of p38α, with Kd = 1.5 nM on p38α. Skepinone-L was effective in cell-free p38αMAPK activity assays with an IC50 of 5 nM, whereas Skepinone-L had an IC50 of 40 nM in whole blood assays. The initial lead compound originally designed by Koeberle et al. was compound 75, which had an IC50 of 0.10 μM for p38α. This scaffold is able to form a tight complementary surface with the linker strand, thereby utilizing small gatekeepers and also inducing l-glycine to flip with carbonyl oxygen. However, due to limited efficacy in vivo, whole blood analysis failed with an IC50 of 3.8 μM. A flexible linear dihydroxypropoxy moiety was later introduced at position 7 of the dibenzocyclic heptenone, as calculations showed that this moiety was well integrated into hydrophobic region II and that its OH group might form additional hydrogen bonds, resulting in the more potent compound Skepinone-L.171 Skepinone-L significantly reduced TNF-α, IL-1β, and IL-10 (IC50 = 30-50 nM). In addition, the production of IL-6, IL-8, IL-13, IL-17α, and IFN-γ was significantly inhibited. However, Skepinone-L did not affect the release of IL-4, IL-5, Monocyte chemoattractant protein-1 and Interferon-gamma inducible protein 10 (high reactive protein). Under its action, LPS-induced TNF-α release was inhibited by 77%,171 which utilizes small gatekeepers with carbonyl oxygen to induce peptide flipping. In addition, an additional hydrogen bond is formed between the terminal OH of the 2, 3-dihydroxypropoxy moiety and the backbone carbonyl group of Gly110, which gives it a better binding force to p38α. Its inhibition of Aβ-induced proinflammatory cytokine production in AD, expression of adhesion proteins associated with cardiac and peripheral inflammation, and induced angiogenesis in cancer have potential therapeutic implications in studies of neurological injury and neurodegeneration after stroke.

Gong et al.172 also designed two promising potential inhibitors 76 and 77 of p38β in 2022, with IC50 of 6.4 and 4.2 nM. During its design, although it is generally believed that p38α and p38β are highly similar, the ATP-binding pocket of p38β is smaller than that of p38α, which could be used to selectively develop p38β inhibitors. Gong et al. optimized the structure based on the known p38α inhibitor Skepinone-L. First, the solvent region of kinase was modified with a hydrophilic group, and the ethylene glycol of Skepinone-L was replaced by a butyric acid group at this position to achieve a significant improvement in the inhibition ability, and the compound 76 was obtained. The IC50 of p38β was 6.4 ± 0.6 nM. Compound 77 was obtained by substitution of a methyl group with a small hydrophobic group in the hydrophobic region II, which further improved the inhibitory ability of p38β with an IC50 of 4.2 ± 0.1 nM. These two optimized compounds have better inhibitory ability than Skepinone-L with excellent inhibitory effect, which is worthy of further in vitro and in vivo experiments.
Sabatini et al.173 developed a series of pyrazolam benzothiazide p38α selective inhibitors in 2015, in which compound 78 had an IC50 of 0.457 μM against p38α and 0.5 μM against TNF-α. In addition, the IC50 of p38β was 1.7 μM, while the inhibitory effect on other kinases such as CDC2, CDK5, JAK2/3, Lck, and SRC was not detected. In its structure with p38α, sulfone oxygen of the ligand interacts with Met109, p-fluorophenyl is located in hydrophobic region I, and the pyrazole ring and Phe169 have stack interactions. In addition, N-2 atoms form anchored hydrogen bonds with Lys53. The low molecular weight, ligand efficiency value and kinase selectivity of pyrazolam benzothiazide compound 78 make this derivative a potentially effective novel inhibitor structure.
In addition, Bartolini et al.174 identified a series of p38 inhibitors with the pyrazo-benzothiazide core in 2019. In addition to compound 78, another different derivative of the same skeleton, COXH11 (79), also showed good p38α inhibitory activity with IC50 = 0.936 μM. Studies in HT29 cells showed that compound 78 had CC50 of 150 μM (24 h) and 95 μM (48 h), whereas COXH11 had no detectable CC50 in cells. They also demonstrated that compounds 78 and 79 inhibited the phosphorylation of p38αMAPK stimulated by LPS and H2O2. The cocrystal structure of the two and p38α (PDB: 5OMG, PDB: 5OMH) shows that the pyridine ring in compound 78 forms a water-mediated contact with the carbonyl group of Phe169 in DFG, the sulfonyl group has a hydrogen bond with the backbone NH of Met109, the fluorophenyl group occupies a hydrophobic pocket, namely hydrophobic region Ⅰ, and the carbonyl group of Phe169 in DFG has a hydrogen bond. Lys53 and Glu71 still had salt Bridges. The conformation of 79 and 78 is significantly different. The hinge contact is achieved by the halide bond between the chlorophenyl group and the His107 carbonyl group. The sulfonyl moiety directly forms a hydrogen bond with the backbone NH of Asp168 in DFG and forms a hydrogen bond with Glu71. In conclusion, compounds 78 and 79 are potent inhibitors of the free radical-dependent pathway activated by p38αMAPK, clearly affecting upstream kinases such as MAP3Ks responsible for the co-activation of p38α MAPK and JNK. All of them can be used as lead compounds. Given their good in vitro activity, their in vivo activity and safety can be further investigated in animal models to provide reference for the development of selective p38 inhibitors for clinical application.
4.2 Inhibitors of p38γ
The inhibitor PIK75 (F7) (80), which selectively inhibits p38γ, was developed for the treatment of cutaneous T-cell lymphomas (CTCL). The enzyme kinetics showed that it was an ATP-competitive p38γ inhibitor with ATPKm of 3.2 ± 0.4 μM and inhibitor Ki of 12.2 ± 1.5 μM, which bound to the ATP-binding pocket of p38γ. PIK75 selectively inhibited p38γ and p38δ with IC50 of 0.028 and 0.055 μM, but had almost no significant effect on p38α and p38β isoforms. In the 3D structural model, PIK75 is predicted to bind to the ATP-binding pocket of p38γ through three hydrogen bonds at Lys56, Tyr59, and Arg70 in the ATP pocket. In the treatment of CTCL, Zhang et al.175 selectively increased p38γ gene expression in CTCL patient samples and cell lines, but not in healthy T cells, suggesting that p38γ plays an important role. The final results showed that pDLGH1Ser158 staining was significantly reduced in PIK75-treated tumors compared with controls. The data indicate that PIK75 reduces cell growth and inhibits p38γ kinase activity both in vitro and in vivo. In addition to inhibiting p38γ, PIK75 also inhibited four isoforms of PI3K with IC50 values of 5.8 nM, 1.3 μM, 76 nM, and 0.51 μM for p110α, β, γ, and δ isoforms. Ultimately, it was shown that inhibition of PI3K p110α alone did not cause CTCL cytotoxicity, whereas inhibition of p38γ caused CTCL cell death.
Pirfenidone (Esbriet, PFD) (81) has been developed for the clinical treatment of idiopathic pulmonary fibrosis, and its inhibitory effect on TNF-α production is one of the important factors for the antifibrosis effect of PFD. Nakazato et al.176 demonstrated that it effectively suppressed the production of proinflammatory cytokines TNF-a, IFN-γ, and IL-6, but enhanced the production of the anti-inflammatory cytokine IL-10. Previous studies have shown that PFD specifically inhibits the kinase activity of p38γ in vitro, but not p38α or p38β isoforms.177 Due to its size and hydrophobicity, PFD is able to diffuse freely across cell membranes without the use of receptors. The compound is rapidly absorbed from the GI tract and reaches its maximum concentration in the blood 1–2 h after oral administration. It is rapidly metabolized at up to 80% in the liver and is metabolized primarily by the hepatic CYP 1A2 enzyme and, to a lesser extent, by other CYP isoforms. Yin et al.178 incubated cancer cells with PFD and found that it blocked p38-γ mediated phosphorylation of several substrates. Including protein-tyrosine Phosphatase H1, estrogen receptor and heat shock Protein 90; several indications suggest that PFD may inhibit AOM/DSS-induced colon cancer development mainly by inhibiting p38γ activity. In addition, it is also a p38γ inhibitor that blocks TGF-β synthesis.179 It can inhibit the expression of p38γ in mice in experiments, but the drawback is that the daily dose of drinking water up to 500 mg/kg of water is required in mice experiments. PFD has been studied clinically as a selective inhibitor of p38γ in several areas, such as a phase III trial for Idiopathic Pulmonary Fibrosi (NCT03208933) conducted between 2017 and 2019. Phase I/II trial of Chronic Kidney Disease (NCT02408744) conducted from 2009 to 2013.
4.3 Inhibitors of p38δ
In the treatment of HNSCC, Sahu et al.180 identified p38δ as a specific target inhibition through the changes of four subtypes of p38 in serum, and designed a small molecule peptide inhibitor WFYH (82) through screening. In the experiments, it was proved that WFYH has a normal binding with three isoforms except p38δ, and p38δ has a Glide score of −12.44, which is the best among the several tetrapeptides listed in the experiments. WFYH has no correlation with Phe169 in p38β and p38γ, but has a hydrophobic interaction with Phe169 in p38α and π-π interaction with p38δ. It is this unique and powerful π-π interaction that causes WFYH to bond well with p38δ. Regarding the selectivity of p38α and p38δ, inhibition of p38α largely requires hydrogen bonding with Met109 and Gly110, and this interaction induces peptide reversal of Met109 and Gly110.181 However, tetrapeptin WFYH forms hydrogen bonds with Gly110, but not with Met109, so it has poor selectivity for p38α. In summary, WFYH is a promising potential p38δ inhibitor. Although no clinical trials have been conducted yet, WFYH can be used as a target for further clinical trials.
SU-002 (83) inhibited kinase activity of p38αT106M in a dose-dependent manner, but no such inhibition was observed in the case of p38α.182 The inhibitory activity of SU-005 (84) on p38γ and p38δ was dose-dependent. IC50 could not be measured due to the insolubility of SU-005 at concentrations over 30 μM. SU-005 impaired the kinase activity of p38γ and p38δ in vitro and in vivo, but not p38α. Although SU-005 showed weak inhibitory activity against preactivated p38γ and p38δ, SU-005 effectively inhibited the automatic phosphorylation of p38γ and p38δ, and SU-005 showed stronger inhibitory activity against labeled p38δ phosphorylation than SU-002.182 Both SU-002 and SU-005 require octadamide or oct-2-acrylamide to inhibit p38γ, so it is speculated that these linked amides may be suitable for shallow hydrophobic sacs formed by Met109 in p38γ. Both p38γ and p38δ are key regulatory factors in the treatment of inflammatory joint destruction in mice. The p38γ or p38δ inhibitor SU-005 identified by Kondoh et al. has potential as a therapeutic agent for RA and diabetes.
The IC50 values of compounds 85, 86, 87, 88 for p38δ were 117 nM, 620 nM, 15.785 μM, and 98.747 μM. Of the four compounds, compounds 85 and 86 can be regarded as nanometer mole inhibitors, while compounds 87 and 88 can be regarded as micron mole inhibitors. Nano mole inhibitors form an additional hydrogen bond (four pairs) with MAPK13 compared to micron mole inhibitors. All four inhibitors form hydrogen bonds with the amido-nitrogen backbone of Met110 (in the hinge) and Asp168 (in the DFG motif). The main difference is the carboxylic acid interaction in the Glu72 side chain. The side chains form hydrogen bonds with linkers between central aromatic and numerous hydrophobic groups in these inhibitors. Nano-mole inhibitors all contain urea junctions, while micro-mole inhibitors all contain amide junctions. In addition, compounds 85 and 86 were in DFG-out binding mode, while 87 and 88 were in DFG-in binding mode. High-efficiency inhibitors were bound to kinase in DFG-out configuration, while low-efficiency inhibitors were bound to kinase in DFG-in configuration. Glide relative butt scores also support the above view.183
AD80 (89) is a type II multikinase inhibitor, which is bound by DFG-out conformation. P38δ and p38γ are direct targets of AD80, and compound 89 is not selective to p38α and p38β. A significant difference among p38 family members is that the gatekeeper residues contain small amounts of threonine for both p38α and p38β at position 106, whereas p38γ and p38δ contain large amounts of methionine at position 109 and 107. It was later proved that AD80 binding is active site-oriented, and the preference for methionine and threonine on gatekeeper may be the cause of its selectivity. AD80 is nearly 100 times more active on liver cancer cells than some of the major FDA-approved small molecule inhibitors for liver cancer (Sorafenib, Lenvatinib, Cabozantinib, etc.). However, in the treatment of hepatocellular carcinoma, forced and sustained MKK6-p38-ATF2 signaling leads to a significant decrease in AD80 activity in hepatocellular carcinoma cells.184
5 PROBLEMS AND CHALLENGES IN THE DEVELOPMENT OF P38MAPK INHIBITORS
Since its discovery many years ago, p38MAPK has been shown to play a key role in many major human diseases, such as cancer, neurodegenerative diseases, inflammation, cardiovascular diseases, and so on. Fortunately, many related inhibitors have been developed and entered into clinical studies (Table 2), but they also face many problems and challenges. The biggest problem is that the current research focuses on the development of inhibitors of p38α and some p38β subtypes, while ignoring p38γ and p38δ subtypes. There are few p38γ and p38δ inhibitors, which may be because p38γ and p38δ lack some regions that p38α and p38β have such as Inhibitor-binding Region. Alternatively, p38α is commonly and heavily expressed in cell lines and tissues, while p38γ and p38δ are tissue-specific, p38γ in skeletal muscle and p38δ in endocrine glands.185
| Name | Target | Phasea | Timea | Diseasea | NCT identifiera |
|---|---|---|---|---|---|
| Compound 19 (TAK-715) | p38α/p38β | Ⅱ | 2004–2005 | RA | NCT00760864 (completed) |
| Compound 25 (PH-797804) | p38α/p38β | Ⅱ | 2008 | RA | NCT00620685 (completed) |
| Ⅱ | 2008 | Neuralgia, postherpetic | NCT00614705 (completed) | ||
| Ⅱ | 2010–2011 | Osteoarthritis | NCT01102660 (completed) | ||
| Ⅱ | 2011–2012 | COPD | NCT01321463 (completed) | ||
| Compound 29 (Losmapimod) | p38α/p38β | Ⅱ | 2014–2016 | Focal segmental glomerulosclerosis | NCT02000440 (completed) |
| Ⅱ | 2014–2016 | COPD | NCT02299375 (completed) | ||
| Ⅱ | 2019–2021 | Facioscapulohumeral muscular dystrophy | NCT04003974 (completed) | ||
| Compound 30 (VX-702) | p38α/p38β | Ⅱ | 2005–2006 | RA | NCT00205478 (completed) |
| Ⅱ | 2006–2007 | RA | NCT00395577 (completed) | ||
| Compound 42 (BIRB-796) | p38α/p38β | Ⅱ | 2001–2002 | Psoriasis | NCT02209753 (completed) |
| Ⅱ | 2001–2002 | RA | NCT02209779 (completed) | ||
| Compound 49 (VX-745) | p38α/p38β | Ⅱ | 2015–2016 | Alzheimer's disease | NCT02423200 (completed) |
| Ⅱ | 2015–2016 | Alzheimer's disease/mild cognitive impairment | NCT02423122 (completed) | ||
| Ⅱ | 2017–2019 | Alzheimer's disease | NCT03402659 (completed) | ||
| Compound 50 (PF-03715455) | p38α/p38β | Ⅰ | 2011 | COPD | NCT01314885 (completed) |
| Compound 56 (Dilmapimod) | p38α/p38β | Ⅰ | 2005–2006 | COPD | NCT00380133 (completed) |
| Ⅰ | 2007 | COPD | NCT00564746 (completed) | ||
| Ⅱ | 2005–2006 | RA | NCT00134693 (completed) | ||
| Ⅱ | 2009–2013 | Acute lung injury | NCT00996840 (completed) | ||
| Compound 58 (SCIO-469) | p38α/p38β | Ⅱ | 2003 | RA | NCT00043732 (completed) |
| Ⅱ | 2004–2006 | Multiple myeloma | NCT00095680 (completed) | ||
| Ⅱ | 2005–2007 | Bone marrow diseases | NCT00113893 (completed) | ||
| Compound 60 (Ralimetinib) | p38α/p38β | Ⅰ | 2016–2017 | Advanced cancer/metastatic cancer/colorectal cancer/nonsmall cell lung cancer | NCT02860780 (completed) |
| Ⅰ/Ⅱ | 2015–2019 | Adult glioblastoma | NCT02364206 (completed) | ||
| Compound 71 (BMS-582949) | p38α/p38β | Ⅰ | 2005–2007 | RA | NCT00162292 (completed) |
| Ⅱ | 2007–2009 | Psoriasis | NCT00399906 (completed) | ||
| Ⅱ | 2008–2010 | Vascular diseases | NCT00570752 (completed) | ||
| Compound 81 (Pirfenidone) | p38γ | Ⅲ | 2017–2019 | Idiopathic pulmonary fibrosis | NCT03208933 (completed) |
| Ⅰ/Ⅱ | 2009–2013 | Chronic kidney disease | NCT02408744 (completed) | ||
| Ⅱ | 2017–2021 | RA interstitial lung disease | NCT02808871 (completed) |
- Abbreviations: COPD, chronic obstructive pulmonary disease; NCT, national clinical trial; p38MAPK, P38 mitogen-activated protein kinase; RA, rheumatoid arthritis.
- a Data were obtained from clinicaltrials.gov (https://clinicaltrials.gov/).
However, recent studies on the important roles of p38γ and p38δ in vivo have shown that the development of inhibitors targeting p38γ and p38δ is very important. For example, p38γ plays a role mainly in the nucleus and inhibits cell migration, which is essential for oncogene-induced senescence and plays an important role in the regulation of cell cycle in cancer cells. Current studies have shown that it is important in breast cancer, squamous cell carcinoma, hepatocellular carcinoma, colorectal cancer, and other cancers.186 However, the role of p38δ in tumor progression cannot be generalized, and it plays a corresponding role in promoting or inhibiting different cancers.187
The essential problem for p38γ inhibitors is that there are conflicting data on p38γ in specific cancer types, and more biological functions of p38γ still need to be further explored, which leads to certain obstacles in the development and application of p38γ inhibitors. Other issues facing the development of p38δ inhibitors include a significant lack of clinical trial evidence, such as compounds 70, 71, 72 have not yet entered clinical trials, and until now no effective and specific p38δ inhibitors have been defined.
P38δ also exists in different cancer types, which may act as a tumor promoter or a tumor suppressor,180, 188 which also leads to difficulties in the development of its specific inhibitors. The structure of compounds that inhibit p38δ also needs to take into account that the isomer has a large quantity of methionine as a gatekeeper residue, which may impair inhibitor binding and selectivity.187
The main problem for the development of p38α and p38β inhibitors is that a series of inhibitors developed in the early stage have failed in vivo clinical trials, such as Pamapimod (5), VX-702 (30), SCIO-323 (55), SCIO-469 (58), and so on. However, some developed inhibitors also have the problem of high toxicity in vivo, such as VX-745 (49). Some of the p38 inhibitors currently in clinical trials are listed below. Most developed p38α and p38β inhibitors have failed in clinical trials due to severe side effects and low safety. Failure may be due to nontarget effects or dose-related toxicity to other kinase or nonkinase targets.189 Many problems have brought some challenges to the development of p38MAPK inhibitors.
6 CONCLUDING REMARKS AND FUTURE PERSPECTIVES
Since its discovery in 1993, p38 MAPK has been studied in an endless stream. A large number of experiments have also proved that it is closely related to many diseases. With the deepening of research, p38MAPK inhibitors have been proved to be important in a variety of tumors, neurodegenerative diseases and other major human diseases. Further studies on the structure and physiological function of p38MAPK have promoted the development of small molecule inhibitors targeting various subtypes of p38MAPK. Currently, many selective p38α inhibitors have been developed, among which Skepinone-L (74) shows excellent inhibitory ability, while other p38γ and p38δ inhibitors are too few to have the most effective comparison significance, and most of them are not specific p38γ and p38δ inhibitors. It can be said that among the existing inhibitors, there is still room for optimization.
First, the optimization of inhibitor design, such as the difference in hinge region rearrangement between MAPK13 and MAPK14, will affect inhibitor binding190 and also have a related impact on inhibitor development. In addition, the development of such allosteric inhibitors is also more important as to which conformation and inhibitor combination is more favorable for different diseases under DFG-in and DFG-out modes.
In addition, drugs may be combined with related enzymes or other inhibitors to treat disease. For example, drugs directly targeting DNA topoisomerase II are an important class of clinical antitumor drugs, and p38γ can actively regulate the signal transduction of topoisomerase II drugs and improve the therapeutic activity of topoisomerase II drugs.191 During this process, p38γ is specifically activated by topoisomerase II targeting drugs, which then activates p38γ to phosphorylate topoisomerase II at Ser-1524, resulting in greater stability and growth inhibition. Therefore, the combined targeting of p38γ and topoisomerase Ⅱ may have more clinical value in tumor diseases.
In recent years, a variety of technical means, such as Affinity Selection Mass Spectrometry, and computer-aided drug design methods have made the design and synthesis of p38MAPK inhibitors more efficient and convenient. These new techniques can be fully utilized in subsequent inhibitor design.
In addition, there are some potential problems, such as the inhibition of p38δ can inhibit tumor progression in tissues, and may also lead to malignant transformation in other tissues; the ability of broad expression of p38α to inhibit disease in specific sites should be considered in future inhibitor studies.
At present, the existing p38MAPK inhibitors have not been accepted by the market. (1) The main problem is organ toxicity. For instance, safety concerns were reported regarding effects on the liver and CNS. One possibility is cross-reaction with other kinases or other cell signaling molecules. Most p38 inhibitors are ATP-competitive inhibitors, and non-ATP-competitive inhibitors cannot in principle have better selectivity.8 The response of ATP-competitive inhibitors and other kinases may lead to failure in clinical trials. In addition, safety concerns are a reason for clinical dose reductions of p38 inhibitors, but specific information about safety concerns is scarce. (2) targeted hepatotoxicity or unexplained treatment-dependent resistance. Elevated liver enzymes have been reported clinically with BIRB-796, VX-745 was discontinued due to undisclosed CNS toxicity in dogs, and people taking SCIO-469 experienced dizziness.192 Based on the limited detailed information associated with these adverse effects, it is not possible to determine whether the problems are pharmacologically or chemically based.
Based on the above problems, several strategies to reduce the toxic side effects in the process of drug development are proposed. (1) The toxic and side effects caused by off-target can be weakened by enhancing the targeting and selectivity of drugs, such as prodrug strategy, antibody-drug conjugate, active tissue targeting via anchored click chemistry (ATTACK), and Proteolysis Targeting Chimera (PROTAC). (2) Lead compound optimization reduces idiosyncratic adverse drug reactions. An important reason for the occurrence of adverse reactions is that the compound has an alert structure, which can be metabolized into active metabolic substances in vivo, triggering cascade reactions and producing toxicity. Therefore, removal or structural modification of alert structures in drugs is an effective way to reduce toxicity. In-depth study of the mechanism of action of drug-induced toxicity so that it can be regulated accordingly to protect organs from less damage.
Finally, considering the important role of p38MAPK in a variety of tumors and the current status of its inhibitors, it is important to develop more efficient and selective p38MAPK inhibitors, PROTACs, and so on. In addition, further understanding of the role of p38MAPK in the body, especially the specific physiological roles of p38γ and p38δ, is needed to provide support for the design of its inhibitors. In addition, the rapid development of precision medicine and biomedical technology in recent decades also provides infinite possibilities for its inhibitors in the future.
AUTHOR CONTRIBUTIONS
Qinwen Zheng: Investigation (lead); resources (lead); writing—original draft (lead). Shutong Li: Resources (equal); writing—original draft (equal). Aoxue Wang: Resources (equal); writing—original draft (equal). Man Zhe: Resources (equal). Panpan Yang: Resources (equal). Yongya Wu: Investigation (equal). Min Zhao: Investigation (equal). Yumeng Zhu: Investigation (equal). Yi Luo: Supervision (equal); writing—review and editing (supporting). Guan Wang: Supervision (equal); funding acquisition (equal); writing—review and editing (supporting). Liang Ouyang: Investigation (equal); supervision (lead); funding acquisition (lead); writing—original draft (equal); writing—review and editing (lead). All authors have read and approved the final manuscript.
ACKNOWLEDGMENTS
This work was supported by grants from the National Natural Science Foundation of China (Grants 82273770, 22177083), Sichuan Science and Technology Program (Grant 2022NSFSC1290), West China Nursing Discipline Development Special Fund Project, Sichuan University (Grant HXHL21011), 1·3·5 project for disciplines of excellence-Clinical Research Incubation Project, West China Hospital, Sichuan University (ZYJC21016). Graphic Abstract and Figure 1 were created by using BioRender.com.
CONFLICT OF INTEREST STATEMENT
The authors declare no conflict of interest.
ETHICS STATEMENT
This study did not involve human participants and/or animals or informed consent. Thus, ethical clearence is not applicable to this article.
Open Research
DATA AVAILABILITY STATEMENT
No data was used for the research described in the article.

