

Prediction of Bond Dissociation Energy for Organic Molecules Based on a Machine-Learning Approach
Yidi Liu
Center of Basic Molecular Science (CBMS), Department of Chemistry, Tsinghua University, Beijing, 100084 China
These authors contributed equally to this work.
Search for more papers by this authorYao Li
Shanghai Institute of Organic Chemistry, University of Chinese Academy of Sciences, Chinese Academy of Sciences, Shanghai, 200032 China
These authors contributed equally to this work.
Search for more papers by this authorQi Yang
Center of Basic Molecular Science (CBMS), Department of Chemistry, Tsinghua University, Beijing, 100084 China
Search for more papers by this authorJin-Dong Yang
Center of Basic Molecular Science (CBMS), Department of Chemistry, Tsinghua University, Beijing, 100084 China
Haihe Laboratory of Sustainable Chemical Transformations, Tianjin, 300192 China
Search for more papers by this authorCorresponding Author
Long Zhang
Center of Basic Molecular Science (CBMS), Department of Chemistry, Tsinghua University, Beijing, 100084 China
Haihe Laboratory of Sustainable Chemical Transformations, Tianjin, 300192 China
E-mail: [email protected]; [email protected]Search for more papers by this authorCorresponding Author
Sanzhong Luo
Center of Basic Molecular Science (CBMS), Department of Chemistry, Tsinghua University, Beijing, 100084 China
Haihe Laboratory of Sustainable Chemical Transformations, Tianjin, 300192 China
E-mail: [email protected]; [email protected]Search for more papers by this authorYidi Liu
Center of Basic Molecular Science (CBMS), Department of Chemistry, Tsinghua University, Beijing, 100084 China
These authors contributed equally to this work.
Search for more papers by this authorYao Li
Shanghai Institute of Organic Chemistry, University of Chinese Academy of Sciences, Chinese Academy of Sciences, Shanghai, 200032 China
These authors contributed equally to this work.
Search for more papers by this authorQi Yang
Center of Basic Molecular Science (CBMS), Department of Chemistry, Tsinghua University, Beijing, 100084 China
Search for more papers by this authorJin-Dong Yang
Center of Basic Molecular Science (CBMS), Department of Chemistry, Tsinghua University, Beijing, 100084 China
Haihe Laboratory of Sustainable Chemical Transformations, Tianjin, 300192 China
Search for more papers by this authorCorresponding Author
Long Zhang
Center of Basic Molecular Science (CBMS), Department of Chemistry, Tsinghua University, Beijing, 100084 China
Haihe Laboratory of Sustainable Chemical Transformations, Tianjin, 300192 China
E-mail: [email protected]; [email protected]Search for more papers by this authorCorresponding Author
Sanzhong Luo
Center of Basic Molecular Science (CBMS), Department of Chemistry, Tsinghua University, Beijing, 100084 China
Haihe Laboratory of Sustainable Chemical Transformations, Tianjin, 300192 China
E-mail: [email protected]; [email protected]Search for more papers by this authorComprehensive Summary
Bond dissociation energy (BDE), which refers to the enthalpy change for the homolysis of a specific covalent bond, is one of the basic thermodynamic properties of molecules. It is very important for understanding chemical reactivities, chemical properties and chemical transformations. Here, a machine learning-based comprehensive BDE prediction model was established based on the iBonD experimental BDE dataset and the calculated BDE dataset by St. John et al. Differential Structural and PhysicOChemical (D-SPOC) descriptors that reflected changes in molecules’ structural and physicochemical features in the process of bond homolysis were designed as input features. The model trained with LightGBM algorithm gave a low mean absolute error (MAE) of 1.03 kcal/mol on the test set. The D-SPOC model could apply to accurate BDE prediction of phenol O—H bonds, uncommon N-SCF3 and O-SCF3 reagents, and β-C—H bonds in enamine intermediates. A fast online prediction platform was constructed based on the D-SPOC model, which could be found at http://isyn.luoszgroup.com/bde_prediction.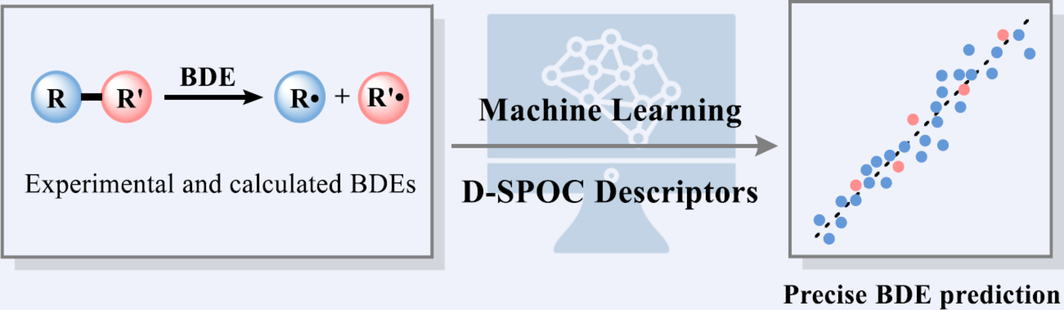
Supporting Information
| Filename | Description |
|---|---|
| cjoc202400049-sup-0001-supinfo.pdfPDF document, 1.2 MB |
Appendix S1: Supporting information |
Please note: The publisher is not responsible for the content or functionality of any supporting information supplied by the authors. Any queries (other than missing content) should be directed to the corresponding author for the article.
References
- 1(a) Blanksby, S. J.; Ellison, G. B. Bond Dissociation Energies of Organic Molecules. Acc. Chem. Res. 2003, 36, 255–263;
(b) Yang, J.-D.; Ji, P.-J.; Xue, X.-S.; Cheng, J.-P. Recent Advances and Advisable Applications of Bond Energetics in Organic Chemistry. J. Am. Chem. Soc. 2018, 140, 8611–8623;
(c) Yang, J.-D.; Xue, J.; Cheng, J.-P. Understanding the role of thermodynamics in catalytic imine reductions. Chem. Soc. Rev. 2019, 48, 2913–2926;
(d) Luo, Y. R. Comprehensive Handbook of Chemical Bond Energies, CRC Press, Boca Raton, FL, 2007;
10.1201/9781420007282 Google Scholar(e) Haynes, W. M. CRC Handbook of Chemistry and Physics, CRC Press, Boca Raton, FL, 2016, p. 96.10.1201/9781315380476 Google Scholar
- 2(a) Brønsted, J. N. Acid and Basic Catalysis. Chem. Rev. 1928, 5, 231–338; (b) Evans, M. G.; Polanyi, N. P. Inertia and driving force of chemical reactions. Trans. Faraday Soc. 1938, 34, 11–24.
- 3(a) Xue, X.-S.; Ji, P.; Zhou, B.; Cheng, J.-P. The Essential Role of Bond Energetics in C-H Activation/Functionalization. Chem. Rev. 2017, 117, 8622–8648, and references cited therein; (b) Feng, Y.; Liu, L.; Wang, J.-T.; Zhao, S.-W.; Guo, Q.-X. Homolytic C−H and N−H Bond Dissociation Energies of Strained Organic Compounds. J. Org. Chem. 2004, 69, 3129–3138; (c) Feng, Y.; Liu, L.; Wang, J.-T.; Huang, H.; Guo, Q.-X. Assessment of Experimental Bond Dissociation Energies Using Composite ab Initio Methods and Evaluation of the Performances of Density Functional Methods in the Calculation of Bond Dissociation Energies. J. Chem. Inf. Comput. Sci. 2003, 43, 2005–2013; (d) St. John, P. C.; Guan, Y.; Kim, Y.; Etz, B. D.; Kim S.; Paton, R. S. Quantum chemical calculations for over 200,000 organic radical species and 40,000 associated closed-shell molecules. Sci Data 2020, 7, 244.
- 4 Bosque, R.; Sales, J. A QSPR Study of O-H Bond Dissociation Energy in Phenols. J. Chem. Inf. Comput. Sci. 2003, 43, 637–642.
- 5 Feng, Y.; Liu, L.; Wang, J.-T.; Zhao, S.-W.; Guo, Q.-X. Homolytic C-H and N-H Bond Dissociation Energies of Strained Organic Compounds. J. Org. Chem. 2004, 69, 3129–3138.
- 6(a) Xue, C. X.; Zhang, R. S.; Liu, H. X.; Yao, X. J.; Liu, M. C.; Hu, Z. D.; Fan, An Accurate QSPR Study of O−H Bond Dissociation Energy in Substituted Phenols Based on Support Vector Machines. J. Chem. Inf. Comput. Sci. 2004, 44, 669–677; (b) Nantasenamat, C.; Isarankura-Na-Ayudhya, C.; Naenna, T.; Prachayasittikul, V. Prediction of Bond Dissociation Enthalpy of Antioxidant Phenols by Support Vector Machine. J. Mol. Graphics Modell. 2008, 27, 188–196; (c) Xu, Q.; Xu, J. Development of validated QSPR models for O–H bond dissociation energy in substituted phenols. Monatsh. Chem. 2017, 148, 645–654.
- 7 Nakajima, M.; Nemoto, T. Machine learning enabling prediction of the bond dissociation enthalpy of hypervalent iodine from SMILES. Sci. Rep. 2021, 11, 20207.
- 8 Qu, X. H.; Latino, D. A.; Aires-de-Sous, J. A big data approach to the ultra-fast prediction of DFT-calculated bond energies. J. Cheminf. 2013, 5, 34.
- 9St. John, P. C.; Guan, Y.; Kim, Y.; Kim, S.; Paton, R. S. Prediction of organic homolytic bond dissociation enthalpies at near chemical accuracy with sub-second computational cost. Nat. Commun. 2020, 11, 2328.
- 10 Wen, M. J.; Blau, S. M.; Spotte-Smith, E. W. C.; Dwaraknath, S.; Persson, K. A. Bondnet: A Graph Neural Network for the Prediction of Bond Dissociation Energies for Charged Molecules. Chem. Sci. 2021, 12, 1858–1868.
- 11(a) Feng, C.; Sharman, E.; Ye, S.; Luo, Y.; Jiang, J. A Neural Network Protocol for Predicting Molecular Bond Energy. Sci. China Chem. 2019, 62, 1698–1703; (b) Yu, H. S.; Wang, Y.; Wang, X. J.; Zhang, J. X.; Ye, S.; Huang, Y.; Luo, Y.; Sharman, E.; Chen, S. L.; Jiang, J. Using Machine Learning to Predict the Dissociation Energy of Organic Carbonyls. J. Phys. Chem. A 2020, 124, 3844–3850.
- 12 Gao, P.; Zhang, J.; Qiu, H.; Zhao, S. A general QSPR protocol for the prediction of atomic/inter-atomic properties: a fragment based graph convolutional neural network (F-GCN). Phys. Chem. Chem. Phys. 2021, 23, 13242–13249.
- 13 Yang, J.-D.; Xue, X.-S.; Ji, P.; Li, X.; Cheng, J.-P. Internet Bond energy Databank (pKa and BDE): iBonD Home Page. http://ibond.nankai.edu.cn.
- 14(a) Yang, Q.; Li, Y.; Yang, J.-D.; Liu, Y.; Zhang, L.; Luo, S.; Cheng, J.-P. Holistic Prediction of pKa in Diverse Solvents Based on Machine Learning Approach Intermediates. Angew. Chem. Int. Ed. 2020, 59, 19282–19291; (b) Yang, Q.; Liu, Y.; Cheng, J.; Li, Y.; Liu, S.; Duan, Y.; Zhang, L.; Luo, S. An Ensemble Structure and Physiochemical (SPOC) Descriptor for Machine-Learning Prediction of Chemical Reaction and Molecular Properties. ChemPhysChem 2022, 23, e202200255; (c) Liu, Y.; Yang, Q.; Cheng, J.; Zhang, L.; Luo, S.; Cheng, J.-P. Prediction of Nucleophilicity and Electrophilicity Based on a Machine-Learning Approach. ChemPhysChem 2023, 24, e202300162.
- 15St. John, P. C., Guan, Y., Kim, Y., Etz, B. D., Kim S.; Paton, R. S. Quantum chemical calculations for over 200,000 organic radical species and 40,000 associated closed-shell molecules. Sci. Data 2020, 7, 244.
- 16 Ke, G.; Meng, Q.; Finley, T.; Wang, T.; Chen, W.; Ma, W.; Ye, Q.; Liu, T.-Y. LightGBM: A Highly Efficient Gradient Boosting Decision Tree. Adv. Neural Inf. Process. Syst. 2017, 30, 3146–3154.
- 17 Zavitsas, A. A. The Relation between Bond Lengths and Dissociation Energies of Carbon−Carbon Bonds. J. Phys. Chem. A 2003, 107, 897–898.
- 18
Bannwarth, C.; Caldeweyher, E.; Ehlert, S.; Hansen, A.; Pracht, P.; Seibert, J.; Spicher, S.; Grimme, S. Extended Tight-Binding Quantum Chemistry Methods. WIREs Comput. Mol. Sci. 2020, 11, e1493.
10.1002/wcms.1493 Google Scholar
- 19 Bergstra, J.; Yamins, D.; Cox, D. D. Making a Science of Model Search: Hyperparameter Optimization in Hundreds of Dimensions for Vision Architectures. In TProc. of the 30th International Conference on Machine Learning (ICML 2013), 2013, pp. I-115 to I-23.
- 20(a) Leroux, F.; Jeschke, P.; Schlosser, M. α-Fluorinated Ethers, Thioethers, and Amines: Anomerically Biased Species. Chem. Rev. 2005, 105, 827–856; (b) Boiko, V. N. Aromatic and heterocyclic perfluoroalkyl sulfides. Methods of preparation. Beilstein J. Org. Chem. 2010, 6, 880–921; (c) Landelle, G.; Panossian, A.; Leroux, F. Trifluoromethyl Ethers and -Thioethers as Tools for Medicinal Chemistry and Drug Discovery. Curr. Top. Med. Chem. 2014, 14, 941–951.
- 21(a) Haas, A.; Moller, G. Preparation and Reactivity of Tris(trifluoromethylselanyl)carbenium [(CF3Se)3C+] and Trifluoromethylsulfanylacetic Acid Derivatives [(CF3S)3–nCXn(O)R]. Chem. Ber. 1996, 129, 1383–1388; (b) Munavalli, S.; Rohrbaugh, D. K.; Rossman, D. I.; Berg, F. J.; Wagner, G. W.; Durst, H. D. Trifluoromethylsulfenylation of Masked Carbonyl Compounds. Synth. Commun. 2000, 30, 2847–2854; (c) Ferry, A.; Billard, T.; Langlois, B. R.; Bacque, E. Synthesis of Trifluoromethanesulfinamidines and -sulfanylamides. J. Org. Chem. 2008, 73, 9362–9365; (d) Xu, C. F.; Ma, B. Q.; Shen, Q. N-Trifluoromethylthiosaccharin: An Easily Accessible, Shelf-Stable, Broadly Applicable Trifluoromethylthiolating Reagent. Angew. Chem. Int. Ed. 2014, 53, 9316–9320; (e) Shao, X.; Xu, C.; Lu, L.; Shen, Q. Structure–Reactivity Relationship of Trifluoromethanesulfenates: Discovery of an Electrophilic Trifluoromethylthiolating Reagent. J. Org. Chem. 2015, 80, 3012–3021; (f) Zhang, H.; Leng, X.; Wan, X.; Shen, Q. (1S)-(−)-N-Trifluoromethylthio-2,10-camphorsultam and its derivatives: easily available, optically pure reagents for asymmetric trifluoromethylthiolation. Org. Chem. Front. 2017, 4, 1051.
- 22 Li, M.; Zhou, B.; Xue, X.-S.; Cheng, J.-P. Establishing the Trifluoromethylthio Radical Donating Abilities of Electrophilic SCF3-Transfer Reagents. J. Org. Chem. 2017, 82, 8697–8702.
- 23(a) Cai, M.; Zhang, R.; Yang, C.; Luo, S. Chin. J. Chem. 2023, 41, 548–559; (b) Li, Y.; Zhang, L.; Luo, S. Bond Energies of Enamines. ACS Omega 2022, 7, 6354–6374.

