

Ligandenaustausch als Weg zu heteroleptischen Carbenhomologen – Synthese des ersten Plumbandiyl-Dimers
Karl W. Klinkhammer
Institut für Anorganische Chemie der Universität, Pfaffenwaldring 55, D-70550 Stuttgart, Telefax: Int.+711/685 4241
Search for more papers by this authorThomas F. Fässler
Laboratorium für Anorganische Chemie, ETH-Zürich, Universitätsstrasse 6, CH-8092 Zürich (Schweiz), Telefax: Int.+1/632 10 90
Search for more papers by this authorHansjörg Grützmacher
Laboratorium für Anorganische Chemie, ETH-Zürich, Universitätsstrasse 6, CH-8092 Zürich (Schweiz), Telefax: Int.+1/632 10 90
Search for more papers by this authorKarl W. Klinkhammer
Institut für Anorganische Chemie der Universität, Pfaffenwaldring 55, D-70550 Stuttgart, Telefax: Int.+711/685 4241
Search for more papers by this authorThomas F. Fässler
Laboratorium für Anorganische Chemie, ETH-Zürich, Universitätsstrasse 6, CH-8092 Zürich (Schweiz), Telefax: Int.+1/632 10 90
Search for more papers by this authorHansjörg Grützmacher
Laboratorium für Anorganische Chemie, ETH-Zürich, Universitätsstrasse 6, CH-8092 Zürich (Schweiz), Telefax: Int.+1/632 10 90
Search for more papers by this authorAbstract
Doppelbindung – ja oder nein? Zentrosymmetrische, trans-gewinkelte Dimere 1 und 2 kristallisieren aus Mischungen der entsprechenden einheitlich substituierten monomeren Carbenanaloga R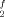 Sn/Sn(SiR3)2 bzw. R
Sn/Sn(SiR3)2 bzw. R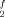 Sn/Pb(SiR3)2. Ob im Plumbandiyl-Dimer 2 eine Doppelbindung vorliegt und damit das letzte Glied aus der Reihe der Ethen-Homologe R4E2 (E = Si, Ge, Pb) hergestellt wurde, muß noch offen bleiben. R = SiMe3, Rf = 2,4,6-(CF3)3C6H2.
Sn/Pb(SiR3)2. Ob im Plumbandiyl-Dimer 2 eine Doppelbindung vorliegt und damit das letzte Glied aus der Reihe der Ethen-Homologe R4E2 (E = Si, Ge, Pb) hergestellt wurde, muß noch offen bleiben. R = SiMe3, Rf = 2,4,6-(CF3)3C6H2.
References
- 1
Aufsätze siehe: a)
G. Rabe,
J. Michl,
Chem. Rev.
1985,
85,
419;
(b)
T. Tsumaraya,
S. A. Batcheller,
S. Masamune,
Angew. Chem.
1991,
103,
916;
10.1002/ange.19911030805 Google ScholarAngew. Chem. 1991, 103, 916; Angew. Chem. Int. Ed. Engl. 1991, 30, 902; (c) M. Drieß, H. Grützmacher, Angew. Chem. Int. Ed. Engl. 1996, 108, 900 bzw.10.1002/ange.19961080805 Google ScholarAngew. Chem. Int. Ed. Engl. 1996, 35, 828; die ersten strukturell charakterisierten Si-S-, Ge-Ge-und Sn-Sn-Doppelbindungssysteme: b) R. West, M. J. Fink, J. Michl, Science 1981, 214, 1343; (e) P. B. Hitchcock, M. F. Lappert, S. J. Miles, A. J. Thorne, J. Chem. Soc. Chem. Commun. 1984, 480; (f) D. E. Goldberg, D. H. Harris, M. F. Lappert, K. M. Thomas, J. Chem. Soc. Chem. Commun. 1976, 261; (g) M. Weidenbruch, H. Killian, K. Peters, H. G. von Schnering, H. Marsmann, Chem. Ber. 1995, 128–973.
- 2(a) H. Grützmacher, H. Pritzkow, F. T. Ebelmann, Organometallics 1991, 10, 23; (b) U. Lay, H. Pritzkow, H. Grützmacher, J. Chem. Soc. Chem. Commun. 1992, 260.
- 3
K. W. Klinkhammer,
W. Schwarz,
Angew. Chem.
1995,
107,
1448;
10.1002/ange.19951071212 Google ScholarAngew. Chem. Int. Ed. Engl. 1995, 34, 1334.
- 4 Ausgewählte physikalische Daten:4: Schmp. (Zers.) 150°C; 1H-NMR (250.133MHz, C6D6, 300K): δ = 0.15 (CH3), 7.88(Harom); 29Si-NMR (39.761 MHz, C6D6, 300K): δ = −5.5 [Si(SiMe3)3], −69.6[Me3Si)3Si];19F-NMR (235.36 MHz, C6D6,200 K); δ−60.5 [ortho-CF3, J (109/107Sn,19F) = 102 Hz, 1J (19F, 13C) = 275.8 Hz], −63.0 [para-CF3, 1J (19F, 13C) = 273.3 Hz]; 119Sn{1H}-NMR (74.631 MHz, C6D6, 300K): 168 (W1/2 = 800 Hz); UV/VIS (n-Pentan): λmax 540 nm. −5: Schmp. (Zers.) ca.90°C; 1H-NMR (250.133 MHz, C6D6, 300K):δ 0.25 (CH3), 8.04 (Hatom); 19F-NMR (235.36 MHz. C6D6, 300 K); δ = −66.6 [ortho-CF3, J(207Pb, 19F) 374Hz, 1J(19F,13C) 275.5Hz]; −62.7 [paraCF3, 1J(19F,13C) 272.5 Hz]; UV/VIS (n-Pentan): λ nm; 1025 nm. Beide Verbindungen ergeben eine korrekte Elementaranalyse.
- 5 Die Durchführung temperaturabhängiger 119Sn-NMR-Mesungen, die Aufschluß über Umlagerungsprozesse oder Anhaltspunkte für ein Monomer-Dimer-Gleichgewicht geben könnten, scheiterte bislang an sehr langen Meßzeiten, der Labilität der Verbindung in Lösung bei Temperaturen oberhalb Raumtemperatur sowie an der schlechten Löslichkeit bei tiefen Temperaturen in inerten Lösungsmitteln.
- 6 G. Trinquier, J. Am. Chem. Soc. 1991, 113, 144.
- 7
Kristallstrukturen: P4 (Siemens), λ(MoKα) = 0.71073 Å, ω-Scan, Lorenzund Polarisationskorrektur, Absorptionskorrektur (ψ-Scan, min/max. Transmission: 0.73/0.99)für ( 5)2, Lösung mit direkten Methoden (SHELXS-86), Verfeinerung mit der Methode der Kleinsten Fehlerquadrate (volle Matrix) gegen F
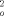 ,(SHELXL-93). ( 4)2. C5H10: monoklin, Raumgruppe C2/ c (Nr. 15), a = 2213.3(4), b = 1530.9(3), c = 2057.1(4) pm, β = 117.99(3)°, V = 6155(2) Å3, Z = 4, (Dimere), ρber. = 1.473Mgm−3, T = −100°C, μ = 1.049mm−1, 3.4° < 2θ < 55°, 7264 gemessene Reflexe, davon 7080 symmetrieunabhängig, 332 Parameter, 22Einschränkungen, R1 [ Fo>4σ ( Fo] = 0.039, wR2 [F
,(SHELXL-93). ( 4)2. C5H10: monoklin, Raumgruppe C2/ c (Nr. 15), a = 2213.3(4), b = 1530.9(3), c = 2057.1(4) pm, β = 117.99(3)°, V = 6155(2) Å3, Z = 4, (Dimere), ρber. = 1.473Mgm−3, T = −100°C, μ = 1.049mm−1, 3.4° < 2θ < 55°, 7264 gemessene Reflexe, davon 7080 symmetrieunabhängig, 332 Parameter, 22Einschränkungen, R1 [ Fo>4σ ( Fo] = 0.039, wR2 [F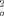 , alle Daten] = 0.110; ( 5)2: monoklin, Raumgruppe I2/a (Nr.15), a = 2164.8(4), b = 945.3(2), c = 2864.2(6)pm, β = 104.87(3)°, V = 5665(2) Å, Z = 4 (Dimere), ρber. = 1.726Mgm−3, T = −100°C, μ = 6.190mm−1, 3.9° < 2θ < 48°, 8675 gemessene Reflexe, davon 4419 symmetrieunabhängig, 289 Parameter, R1 [ Fo>4σ( Fo)] = 0.038, wR2 [ F
, alle Daten] = 0.110; ( 5)2: monoklin, Raumgruppe I2/a (Nr.15), a = 2164.8(4), b = 945.3(2), c = 2864.2(6)pm, β = 104.87(3)°, V = 5665(2) Å, Z = 4 (Dimere), ρber. = 1.726Mgm−3, T = −100°C, μ = 6.190mm−1, 3.9° < 2θ < 48°, 8675 gemessene Reflexe, davon 4419 symmetrieunabhängig, 289 Parameter, R1 [ Fo>4σ( Fo)] = 0.038, wR2 [ F , alle Daten] = 0.072. Die kristallographischen Daten (ohne Strukturfaktoren) der in dieser Veröffentlichung beschriebenen Strukturen wurden als „supplementary publication”︁ no. CCDC-100326 beim Cambridge Crystallographic Data Centre hinterlegt. Kopien der Daten können kostenlos bei folgender Adresse in Großbritannien angefordert werder: The Director, CCDC, 12 Union Road, Cambridge CB2 1EZ (Telefax: Int. +1223/336-033; E-mail: [email protected]).
, alle Daten] = 0.072. Die kristallographischen Daten (ohne Strukturfaktoren) der in dieser Veröffentlichung beschriebenen Strukturen wurden als „supplementary publication”︁ no. CCDC-100326 beim Cambridge Crystallographic Data Centre hinterlegt. Kopien der Daten können kostenlos bei folgender Adresse in Großbritannien angefordert werder: The Director, CCDC, 12 Union Road, Cambridge CB2 1EZ (Telefax: Int. +1223/336-033; E-mail: [email protected]).
- 8 H. Jacobsen, T. Ziegler, J. Am. Chem. Soc. 1994, 116, 3667.
- 9 Rechnungen: Gleichgewichtsstrukturen von E2H4 wurden mit CCD(Sn) und CCSD-Rechnungen (Pb) (Gaussian 94) mit den Stuttgarter quasi-relativistischen Pseudopotentialen [W. Kuechle, M. Dolg, H. Stoll, H. Preuss, J. Chem. Phys. 1991, 74, 1245] und erweiterten Basissätzen für Sn und Pb: (4s4p2d)/[5s5p2d] bestimmt; Basisansatz für H: (3s2p)/[4s2p] [T. H. Dunning, J. Chem. Phys. 1970, 19, 553]. Die Dimerisie-rungsenergien wurden auf CCSD-Niveau unter Berücksichtigung von Basissatz-Superpositionsfehlern (Full-Counterpoise-Methode) und Nullpunktsenergien berechnet. Lokale Minima auf der Hyperfläche wurden durch Frequenzanalysen (NIMAG = 0) abgesichert.
- 10 Quantenmechanisch läßt sich eine Doppelbindung über das Vorhandensein zweier gemeinsamer Elektronenpaare je Atompaar, also unabhängig von der Topologie oder der inhärenten Bindungsenergie, definieren. Gängige Methoden-im Rahmen der LCAO-OM-Näherung (LCAO = Linear Combination of Atomic Orbitals)- sind beispielsweise die NBO- oder die NLMO-Analyse (NBO = Natural Bond Orditals, NLMO = Naturally Localized Molecular Orbitals) [J. E. Carpenter, F. Wienhold, J. Mol. Struct. THEOCHEM 1988, 169, 41]. Außerhalb dieser Näherung ist-als Funktion der kinetischen Energie der Elektronen- die Elektronen-Lokalisierung-Funktion (ELF) zu nennen [ B. Silvi, A. Savin, Nature 1994, 371, 683]. über eine detaillierte quantenchemische Analyse der Bindungsverhältnisse in Verbindungen wie ( 5)2 wird von uns an anderer Stelle berichtet werden.
Citing Literature

This is the
German version
of Angewandte Chemie.
Note for articles published since 1962:
Do not cite this version alone.
Take me to the International Edition version with citable page numbers, DOI, and citation export.
We apologize for the inconvenience.
