

On the diversity of F420-dependent oxidoreductases: A sequence- and structure-based classification
Funding information: Dutch Research Council NWO VICI-grant; European Union's Horizon 2020 research and innovation programme, Grant/Award Number: 847675; Fondo para la Investigación Científica y Tecnológica, Grant/Award Number: PICT 2016-2839
Abstract
The F420 deazaflavin cofactor is an intriguing molecule as it structurally resembles the canonical flavin cofactor, although behaves as a nicotinamide cofactor due to its obligate hydride-transfer reactivity and similar low redox potential. Since its discovery, numerous enzymes relying on it have been described. The known deazaflavoproteins are taxonomically restricted to Archaea and Bacteria. The biochemistry of the deazaflavoenzymes is diverse and they exhibit great structural variability. In this study a thorough sequence and structural homology evolutionary analysis was performed in order to generate an overarching classification of the F420-dependent oxidoreductases. Five different deazaflavoenzyme Classes (I–V) are described according to their structural folds as follows: Class I encompassing the TIM-barrel F420-dependent enzymes; Class II including the Rossmann fold F420-dependent enzymes; Class III comprising the β-roll F420-dependent enzymes; Class IV which exclusively gathers the SH3 barrel F420-dependent enzymes and Class V including the three layer ββα sandwich F420-dependent enzymes. This classification provides a framework for the identification and biochemical characterization of novel deazaflavoenzymes.
1 INTRODUCTION
F420 is a naturally occurring deazaflavin cofactor in which the N5 atom of the isoalloxazine ring is substituted by a carbon atom and has an 8-hydroxyl moiety instead of the methyl groups at positions 7 and 8, compared to the canonical flavin cofactors FMN and FAD. It was first isolated almost 50 years ago by Cheeseman et al.1 and its structure was solved shortly after.2 F420 is an obligate two-electron hydride carrier with a low standard redox potential (−340 mV), which resembles that of nicotinamide cofactors (−320 mV) rather than that of flavins (−220/−190 mV).3 The first reports on F420-dependent enzymes were related to methanogenesis in archaeal species.4, 5 For a long time these were considered as unusual redox enzymes. Recent research has demonstrated that deazaflavoenzymes are actually widespread across Archaea and Bacteria.3 In Archaea, the species from the euryarchaeota phyla Methanosarcina spp, Methanothermobacter spp and Archaeoglobus fulgidus are among the most frequently investigated. In Bacteria, research has been centered on the Actinobacteria phylum including Mycobacterium, Streptomyces and Nocardia genera.3 The restricted domain distribution of the F420 cofactor and its connection to anaerobic metabolism, highlight its status as a relic from the origin-of-life world.3
Previously, a number of F420-dependent enzymes have been classified according to their three-dimensional fold into three groups: the luciferase-like hydride transferases (LLHTs) (also known as luciferase-like monooxygenases, LLMs), the pyridoxamine-5′-phosphate oxidases (PNPOxs), and the deazafavin-dependent nitroreductases (DDNs).6 Although this classification was based on structural homology, it should be noted that the PNPOx and DDN representatives display the same split β-barrel fold. While all known DDNs exclusively rely on F420, LLHT and PNPOx members show dependence on other flavin cofactors as well.7, 8 Recently, the aflatoxin degrading F420-dependent reductases from actinomycetales were discovered and characterized.9 These were described as F420-dependent reductases (FDRs A and B) and are homologous to members of the PNPOx family.10 Later, it was proposed that FDRs should be instead referred to as flavin/deazaflavin oxidoreductases (FDORs A and B). The FDOR-A group exclusively includes F420-dependent enzymes, while FDOR-B encompasses deazaflavoenzymes as well as enzymes using FMN, FAD, and heme cofactors.11 Except for those utilizing F420H2 in the reduction of metabolites, there are many known enzymes that use the deazaflavin cofactor for other reactions. Among them are oxidoreductases that can shuttle a hydride between nicotinamide and F420 (FNOs),12, 13 oxidases that use F420 coupled to FMN to reduce dioxygen (FprA),14 and dehydrogenases that employ reduced F420 to reduce the methenyl-H4MPT+ cofactor.15 Additionally, other redox enzymes in anaerobic metabolism such as [NiFe]-hydrogenases16 and thioredoxin reductases17 depend on the F420 cofactor and show unique structural features. Therefore, considering the increasing number of characterized F420-dependent enzymes displaying not only different biochemistries but also a variety of structural topologies, a structure-based classification of deazaflavoproteins would be valuable.
Enzyme classification can be performed either on the basis of functionality (e.g., the chemical reaction they catalyze18, 19) or on the basis of the evolutionary history,20 among other criteria. One of the main drawbacks of the first strategy is that it relies on features lacking a common evolutionary origin. Although these phenetic classifications can be a very useful way of organizing protein information, problems arise when investigating the underlying determinants of enzyme functionality. Classifications based on observable traits, regardless of the phylogeny, also perform poorly in predicting the activity of newly found enzymes, especially when the number of items to classify increases constantly over time.18 Molecular evolution analysis allows for understanding how modern enzymes work.21 The historical and physical causes of a protein function can be traced through their trajectory until its emergence. In this way, the determinants of enzyme function will become evident. Therefore, the evolutionary-based classifications are a robust approach to infer the activity of new enzymes and to explore its physico-chemical and biological determinants. Several examples are available in the literature with CATH22 and pfam23 as gold standards.
In this work the oxidoreductases that use the deazaflavin cofactor F420 were comprehensively analyzed from the structural homology perspective. Five different classes were defined that encompass the whole enzymatic and structural diversity of F420-dependent oxidoreductases recognized at the moment. Besides, the phylogenetic analyses of these classes are reported and the trends in the cofactor utilization are analyzed.
2 METHODS
2.1 Evolutionary clustering of F420-dependent enzymes
Sequences from F420-dependent enzymes for which structures have been obtained were collected from PDBsum (PDBsum, RRID:SCR_006511). Domain architecture was investigated using the hierarchical criteria from CATH (CATH: Protein Structure Classification, RRID:SCR_007583) and/or pfam (Pfam, RRID:SCR_004726). InterPro and TIGRFAMs databases were also consulted (InterPro, RRID:SCR_006695; JCVI TIGRFAMS, RRID:SCR_005493) (last accession to databases: December 2020–January 2021). Structures were clustered according to their domain topology and architecture as the definitive criteria for a shared evolutionary origin. Structural alignment of each group was performed in PROMALS3D (PROMALS3D, RRID:SCR_018161).
2.2 F420-dependent oxidoreductases class profiling
For each structural superfamily identified, sequence datasets were constructed by homology searches using as queries the sequences of the enzymes retrieved from structural databases (vide supra). Blastp (BLASTP, RRID:SCR_001010) was conducted using non-redundant protein sequences database and specifying the taxonomy (Archaea or Bacteria). The 250/500 first hits were collected on each search (E-value ≤1e-9). HMM profiling was conducted in HMMER (Hmmer, RRID:SCR_005305) using UniprotKB database and the first 250/500 hits were collected. Enzymes sharing the same structural domain but not using the F420 cofactor were also used as queries in the homology searches and the retrieved sequences were included in the datasets to ensure the clustering was not biased by the data collection. For Class II, pfam available datasets were collected in complete form as well. Raw datasets contained >1000 seqs for each superfamily. MAFFT v7 (MAFFT, RRID:SCR_011811) was employed to build multiple sequence alignments (MSAs). Redundancy was removed (cut-off = 80% identity) with CD-HIT (CD-HIT, RRID:SCR_007105). MSAs were visually inspected and single sequence insertions/extensions trimmed. Substitution models and alignment parameters were calculated in ProtTest 3.4.2 (ProtTest, RRID:SCR_014628). Phylogenies were constructed by the maximum likelihood inference method implemented in PhyML 3.1 (PhyML, RRID:SCR_014629) or RaxML 8.2.12 (RAxML, RRID:SCR_006086) with 100/500 bootstraps (BS), respectively. Transfer bootstrap values (TBE) were obtained in BOOSTER.24 For Class III, Bayesian inference was also conducted in Mr Bayes 3.2.6 (MrBayes, RRID:SCR_012067) running 2000000 generations until convergence <0.2 was reached. FigTree 1.4.2 (FigTree, RRID:SCR_008515) was employed to visualize and edit the trees. HMM logos were created with the WebLogo 3 online server (WEBLOGO, RRID:SCR_010236) for sequences exclusively using F420 according to the obtained phylogenies. Sites in contact with F420 were retrieved from the PDB structures employing the SAS server (SAS—Sequence Annotated by Structure, RRID:SCR_004635).
3 RESULTS AND DISCUSSION
A molecular evolution analysis was conducted with the aim to integrate present biochemical knowledge on F420-dependent enzymes. Currently known enzymes that use F420/F420H2 as cofactor in oxidation/reduction processes were included. A shared structural fold was considered as the first sign of a common evolutionary origin.20
Initially, the strategy consisted of mining the structural databases to retrieve all enzymes using F420 as a cofactor. The 50 collected structures (out of 171718 PDB entries scanned), belong to 24 different deazaflavin oxidoreductases. By analyzing the domain topology and architecture, these enzymes were assigned to four different evolutionary-independent units. In that way, four different unrelated folds were identified among the gathered structures (Classes I–IV). For those enzymes for which no structures are available, but confirmed to depend on F420,17 a second sequence dataset was built and the domain topology predicted with GENE3D/CATH.22 From this analysis, one extra distinct fold was identified (Class V). Structure-based alignments were constructed when possible for each of the identified classes and the evolutionary relationships were inferred. Next, homology searches and HMM profiling were employed to find close and distant homologs and sequence-based phylogenies were constructed (see Section 2). All the evolutionary and structural information obtained was integrated with the available biochemical data.
This step-wise analysis allowed us to propose the existence of five well-defined Classes (I–V) of deazaflavoenzymes (Tables 1 and S1). Classes are defined by the structural superfamily to which the sequences belong. For some of the classes, different types of enzymes are proposed on the basis of their phylogenetic clustering and their shared catalytic properties. These will be described in detail in the coming sections.
| Class/Type | Structural superfamily | Domain | Archetypal examples | Biotechnological applications | Ref | ||||
|---|---|---|---|---|---|---|---|---|---|
| Name | Function | Electron flowa | Ref | ||||||
| I | A | TIM barrel | 
|
tMer | F420-dependent N5,N10-methylene-H4MPT reductase | 
|
25 |
|
26-29 |
| B | Fgd | F420-dependent glucose-6-phosphate dehydrogenase | 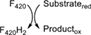
|
30 | |||||
| Adf | F420-dependent secondary alcohol dehydrogenase | 31 | |||||||
| C | LmbY | Apd6 (4-alkyl-proline derivative) F420-dependent reductase | 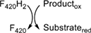
|
32 | nd | - | |||
| II | A | Rossmann fold | 
|
Fno | F420H2:NADP+ oxidoreductase | 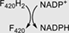
|
33 |
|
28 |
| B | 
|
Mtd | F420-dependent methylene-H4MPT dehydrogenase | 
|
15 | ||||
| C | 
|
Fpra | F420H2 oxidase | 
|
34 | ||||
| III | β-roll | 
|
Ddn | Deazaflavin-dependent nitroreductase | 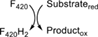
|
35 |
|
26, 27, 29 | |
| Fdr | F420H2-dependent reductase | 28 | |||||||
| IV | SH3 barrel | 
|
FrhB | F420-reducing [NiFe]-hydrogenase, subunit beta | 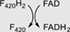
|
16 | nd | - | |
| V | 3-layer ββα sandwich | 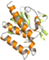 b b
|
DFTR | F420-dependent thioredoxin reductase | 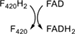
|
17 | nd | - | |
- Note: Folds are colored according to secondary structure elements representing CATH hierarchical classification. Presented domains: Class I: 3b4yA00, Class II: 1jaxA00 (FNO), 1qv9A00 (MTD), 2ohhA02 (oxidases), Class III: 3r5rA00, Class IV: 4omfB02, Class V: 1trbA01. Extended information is available in Table S1.
- Abbreviation: nd, not determined.
- a The physiological reaction is indicated, while the reverse reaction is typically also feasible.
- b Selected by homology (E = 5.5e–16).
3.1 Class I: TIM barrel F420-dependent enzymes
This class is defined by the conserved α/β barrel fold and includes the so-called LLHTs (also referred as LLMs). Two kinds of well-known F420-dependent oxidoreductases are found here: the methylene-H4MPT reductases (MERs)25 and the dehydrogenases, represented by the F420-dependent glucose-6-phosphate dehydrogenases (FGDs) (Table 1).30 Depending on the physiological conditions, MERs can consume or produce F420H2.36 FGDs essentially generate F420H2, although the reversibility of their activity has also been demonstrated.31 Also, other enzymes are found in this group which are involved in antibiotics biosynthesis37 or in the production of pathogenesis related molecules such as outer membrane lipids like the phthiocerol dimycocerosates.38 Hitherto, biochemical details of these other members are scarce. Recently, Steiningerova et al.32 demonstrated that the actinobacterial enzymes LmbY,39 HrmD,40 among others, are F420H2-dependent reductases displaying the TIM barrel fold. These enzymes are involved in the last step of 4-alkyl-l-proline derivatives (APDs) biosynthesis. The APD moiety is common among structurally and functionally different metabolites.41
Owing to the enzymatic diversity found among this class, a structure-based phylogeny was constructed to unveil the deep evolutionary relationships among the different members (Figure S1A). Two F420-dependent enzyme clusters can be discerned, one including the MERs and FGDs, while the other including the actinobacterial F420 reductases along with bacterial FMN-dependent enzymes such as the alkanesufonate monooxygenase (SsuD, PDB: 1m41)42 and the nitrilotriacetate monooxygenase (NTA_MO, PDB: 3sdo). This distribution was further confirmed by inferring the sequence-based phylogeny employing a robust and representative dataset (Figures 1 and S1B). Among the first cluster of deazaflavoenzymes (TBE/PP = 0.94/0.99), two well-defined clades of F420-dependent enzymes are observed. These will be designated here as Type A and Type B, formerly referred to as reductases and dehydrogenases clades.29, 43 Type A includes the MERs, the mycobacterial phthiodiolone ketoreductase fPKR38 and the reductase LxmJ from the lexapeptide gene cluster found in Streptomyces rochei37 (TBE/PP = 0.92/0.99). The Type B cluster is populated by the FGDs, the F420-dependent sugar-6-phosphate dehydrogenases (FSDs), and the alcohol dehydrogenases: hydroxymycolic acid dehydrogenase (fHMAD) from M. tuberculosis44 and secondary-alcohol dehydrogenase Adf (PDB: 1rhc) from Methanoculleus thermophilicus31 (TBE/PP = 0.99/1). The other cluster of deazaflavoenzymes identified in the structural phylogeny, seems to derive from FMN-dependent sequences with good support (TBE/PP = 0.92/0.74). This clade is designated Type C and includes the F420-dependent reductases encoded within the actinobacterial biosynthetic gene clusters of APDs (called Apd6 proteins based on the catalyzed step in the pathway).45 Among the FMN-dependent homologs the well-known bacterial FMN-dependent luciferases (e.g., LuxB, PDB: 1luc)46 are found.

The unveiled topology allows proposing two independent origins for the F420-dependent enzymes within this class. While Type A and B are early diverging from a sister clade of FMN-dependent sequences, the Type C is branching out from a clade of FMN-dependent enzymes at a posterior stage. The taxonomic distribution shows a mixed arrangement of bacterial and archaeal species with predominance of facultative anaerobes and methanogens for Types A and B. In the case of the Type C cluster, actinobacterial sequences are found exclusively, indicating a very restricted taxonomic distribution. Overall, the topology of the tree suggests that cofactor specificity (FMN or F420) has played a major role as selective pressure in the evolutionary history of members of this class. Besides, the emergence of Type C is strongly linked to the Actinobacteria phylum. Finally, it is important to note that several Type A and B enzymes have been reported to display suitable activities for biotechnological applications as recently reviewed by Shah et al.29 (Table 1). In the case of Type C, as these are a newly characterized group, their biochemistry and biotechnological potential awaits exploration.
3.2 Class II: Rossmann fold F420-dependent enzymes
Class II members are deazaflavoenzymes that share the classic 3-layer αβα sandwich topology, commonly known as Rossmann fold. Members with known biochemical features vary considerably in structure and function. These are the F420H2-NADP+ oxidoreductases (FNOs),33 the F420-dependent methylene-H4MPT dehydrogenases (MTDs),15 and the F420H2 oxidases (FprAs).34 The latter show a unique multidomain architecture, as they are N-terminally fused to a ~180-amino acid β-lactamase domain (PF00753, CATH 3.60.15.10). Despite the structural differences the catalytic strategy is similar, as the hydride transfer proceeds from F420H2 to a second cofactor such as a nicotinamide (FNOs), tetrahydromethanopterin (MTDs) or a flavin (FprAs)34 (Table 1).
In spite of some structural variations of the central common fold, a structure-based phylogeny could be constructed (Figure S2A). MTDs and oxidases emerge as clear monophyletic groups (BS = 97 and BS = 100, respectively). FNOs, although sharing ancestry, form a poorly supported paraphyletic group. Considering the topology of the structural phylogeny plus the sequence similarity among each group, we propose the existence of three types of deazaflavoenzymes inside this class. These are designated Type A, Type B and Type C (Table 1). Phylogenies were constructed for each of them, revealing some shared and other unique evolutionary paths (Figure 2).

The Type A members are the known F420H2-NADP+ oxidoreductases. Phylogenetic analysis shows that all members of this subclass derive from a single ancestor (TBE = 0.82) and are distributed in two groups (Figures 2 and S2B). This clear splitting does not respond to any taxonomic or structural feature and thus should be further investigated. The FNO-type sequences are 200-220 amino acids in length, show single domain architecture (PF03807, CATH 3.40.50.720) and use F420 and NADP+. FNOs are homologous to some NAD(P)H-dependent reductases which encode for an extra C-terminal domain of 40–50 amino acids in length. These reductases are early diverging in the phylogeny and do not use a deazaflavin cofactor (Figure 2). Among them, the NAD(P)H-dependent pyrroline-5-carboxylate reductases, forming a monophyletic group of 5 sequences (TBE = 0.95),47 and the D-apionate oxidoisomerase (apnO, PDB: 5t57)48 are found (Figure S2B). The topology of the tree strongly suggests that the use of nicotinamide cofactor is the ancestral feature, whereas the utilization of F420 arose later in evolution. Taxonomic distribution of Type A enzymes seems to be enriched in anaerobic or facultative anaerobic species either from Bacteria or Archaea.
Type B includes the known F420-dependent methylenetetrahydromethanopterin dehydrogenases (Figures 2 and S2D). This type is composed solely of F420-using enzymes that display an exclusive structural domain (PF01993, CATH 30.40.50.10830). The taxonomic distribution is strongly biased toward methanogenic archaea belonging to the euryarchaeota phylum.15, 49 These enzymes are also present in Archaeoglobi species50 which, despite being sulfur-metabolizing organisms, encode a nearly complete set of genes for methanogenesis.51 A few eubacterial-derived proteins are also observed. However, none of these sequences have been experimentally characterized.
Type C, including the F420H2 oxidases (FprA), shows another different history (Figures 2 and S2F). The two-domain architecture (lactamase B [pfam 00753]- flavodoxin [pfam 00253]) defining this group is shared by various other redox enzymes such as nitric oxide reductases52 and flavodiiron proteins.53 The evident difference among these FMN-dependent enzymes is the electron donor preference, which can be F420H2 or NADH. For those working as nitric oxide reductases no other cofactor than FMN is required. Phylogenetic analysis shows that F420 usage is restricted to one particular clade (TBE = 1) containing all known FprAs, embedded into a large group of FMN-dependent enzymes (Figures 2 and S2F). On the other hand, those enzymes reportedly relying on NADH as electron donor form a monophyletic group (TBE = 1) which has a sister clade of uncharacterized homologs inferred to be FMN-dependent enzymes. In that context, similar as observed for Class I, the usage of FMN seems to be the ancestral feature, while by two later independent events F420H2 and NADH specificities arose. Remarkably, the utilization of F420 by Type C members seems to be restricted to archaeal species,14, 34 while NADH is used by bacteria.53, 54 However, this statement should be considered cautiously as very few members of the family have been experimentally characterized.
3.3 Class III: β-roll F420-dependent enzymes
This class encompasses F420-dependent reductases of great biochemical diversity: among them the well-known DDNs35 and the F420H2-dependent reductases,10 also referred to as FDOR A and B.11 All enzymes display an arrangement of antiparallel beta sheets commonly called split β-barrel fold. The well-known FMN-dependent PNPOxs8 and other FAD and heme-dependent enzymes show sequence similarity to proteins in this group. A thorough sequence and structural analysis of the mycobacterial deazaflavoenzymes belonging to this class was already reported by Ahmed et al.11 In this work we explore the whole taxonomical diversity of the split β-barrel reductases to disclose general evolutionary trends and thus to provide a robust description of the class.
Our global analysis of the available protein structures allowed us to construct a structure-based phylogeny with the purpose of unveiling if clustering corresponds to cofactor specificity. The topology of the obtained tree revealed that the structural superfamily has no structurally imposed cofactor preference, as an interleaved distribution is observed (Figure S3A). This was further confirmed by a sequence-based phylogenetic analysis which resulted in the same clade distribution. By constructing a robust dataset -via ensuring diverse taxonomic representation and lowering redundancy- the scattered cofactor dependence became clear (Figures 3 and S3B). Hence, Class III deazaflavoenzymes are mixed with enzymes showing different cofactor preferences. The tree shows some hard polytomies even after inferring the phylogeny by Maximum Likelihood and Bayesian methods. However, well-supported clades could be identified and some trends in cofactor usage became clear. These are discussed below.

All sequences previously characterized as FDOR-As11 (MSMEG_3660, 2027, 3004, 3356 and Rv3547), sharing the use of F420 molecule as cofactor, form a monophyletic group (TBE/PP = 0.96/0.75). However, the cofactor preference for F420, FMN, FAD and heme is interspersed in the rest of the tree. It becomes evident that the FDOR-B members11 do not form a monophyletic group. The clade comprising the aflatoxin degrading enzyme MSMEG_3380, an FDOR-B, includes the early diverging FMN-dependent enzymes Npun_R6570 from Nostoc punctiforme and MSMEG_5675 with very good confidence (TBE = 0.83). Similarly, a distinct clade is observed containing the FDOR-Bs rv1155, MSMEG_5170, rv2074, rv2991 and the heme-dependent enzyme MSMEG_6519 (TBE = 0.78). This irregular distribution becomes even more evident in the clade including the F420-dependent MSMEG_652611 and the heme storage protein HutZ (PDB: 3tgv) (TBE = 0.92). Among this clade, an early diverging group of reductases displaying various cofactor specificities is found. These are the NimA/NimC protein from Clostridium acetobutylicum, the FDOR-B MSMEG_5243, the F420-dependent dehydropiperidine reductase TpnL involved in the thiopeptins biosynthesis,55 the FAD-dependent reductase component of the aromatic hydroxylases pheA2 and TTHA0961,56, 57 and the FMN-dependent small reductase component of a styrene monooxygenase TthH11_19250.58 From this analysis, it is evident that cofactor preference is not the trait selected during the evolution of this class, as cofactor specificity is mixed. This also echoes in the flexibility of the cofactor binding site, which allows a certain degree of promiscuity as some reductases seem to be able to utilize both F420 and FMN.59 This interspersed distribution imposes implications for gene annotation protocols and applied enzymology, as cofactor specificity of new enzymes can be hardly predicted from sequence and/or structure for structurally related enzymes to this class. Instead, it should be experimentally determined as it has been previously shown.11 The biotechnological application of members of this class is a promising ground as it has been reviewed by others3, 29 (Table 1). Finally, it should be noted that although most of the research has been devoted to Mycobacterium species, proteins from other bacteria as well as Archaea are observed among all clades (Figure S3B).
3.4 Class IV: SH3 barrel F420-dependent enzymes
Class IV is formed exclusively by deazaflavoenzymes displaying on their structure the small β-barrel domain (≈60 amino acids) consisting on five/six β-strands arranged as two tightly packed anti-parallel β sheets. This F420-binding domain is found in the F420-reducing [NiFe]-hydrogenases, a complex of three subunits (FhrABG) involved in the oxidation/reduction of F420 in the methanogenesis pathway in Archaea.16, 60-62 Specifically, subunit B (FhrB) contains a FAD molecule, a [4Fe-4S] center and the F420 binding site. It displays the distinctive SH3 barrel domain (PF04422) at the N-terminal region and the PF04432 at the C-terminus. This subunit is devoted to the reduction of F420 at the expense of electrons transferred from a flavin/ferredoxin system (Table 1). Despite the lack of many 3D structures, a few homologs have been characterized in detail. When analyzing their predicted topology they all display the same structural domain. The F420-reducing [NiFe]-hydrogenases, Fpo-F63 and Fhr-B,61 the F420-dependent sulfite dehydrogenase (FSR)64 and the Hdr (heterodisulfide reductase)65 populate this class. All sequences in this Class belong exclusively to the Archaea domain (Figures 4 and S4A).

3.5 Class V: 3-layer ββα sandwich F420-dependent enzymes
Class V has a single biochemically characterized member with no solved 3D structure, namely the deazaflavin-dependent FAD-containing thioredoxin reductase (DFTR) from Methanocaldococcus jannaschii (Mj-TrxR).17 This enzyme has a predicted fold similar to the NAD(P)H-dependent FAD-containing thioredoxin reductases (NTR) consisting of a three-layer sandwich. However, the classic nicotinamide cofactor is replaced by F420H2 as the electron donor to reduce the bound FAD (Table 1). Our sequence analysis does not reveal any unique feature accounting for such a change in the cofactor specificity, rather than a few changes in the predicted NAD(P)H binding regions. From the evolutionary history reconstructed by Susanti et al.,17 it seems clear that DFTR belongs to the disulfide oxidoreductase family of flavoenzymes. However, as the majority of the close homologs have not been experimentally characterized, it is hard, if not impossible, to make assumptions on the electron donor specific distribution across the superfamily. It has been proposed that DFTRs would only be present in the methanococci Archaea class replacing the NTRs.66 If that were the case, the utilization of F420H2 may be a derived feature of the protein family. However, a deeper understanding of the coenzyme dependence of members of this class is strongly contingent on the discovery of new deazaflavoenzymes displaying this fold.
4 CONCLUSION
A global classification of the oxidoreductases that employ the deazaflavin F420 cofactor is proposed on the basis of the evolutionary analysis of available sequences and structures. This classification may facilitate annotation of deazaflavoenzyme-encoding genes and allow identifying novel F420-dependent enzymes. Besides, it can help to disclose the origin and extent of the functionalities associated with the F420 cofactor. Furthermore, this study provides clues on how F420-dependent enzymes are evolutionarily related to other redox enzymes that rely on cofactors like heme, flavin, and nicotinamide.
ACKNOWLEDGMENTS
This work was supported by the COFUND project oLife that has received funding from the European Union's Horizon 2020 research and innovation programme under grant agreement No 847675, Agencia Nacional de Promoción Científica y Tecnológica (ANPCyT) (PICT 2016-2839 to M. L. M.) and the Dutch Research Council NWO (VICI-grant to M. W. F.). The authors declare no conflict of interest.
Open Research
DATA AVAILABILITY STATEMENT
The data that supports the findings of this study are available in the supplementary material of this article.

