

Engineering nanostructured pure cancer cell membrane-derived vesicles as a novel therapeutic cancer vaccine
Hongliang He and Chunqing Guo contributed equally to this study.
Abstract
Extracted cancer cell membrane carries the antigens of the parent tumor cell. This autologous antigen repertoire presents cancer cell membrane-derived nanoparticles highly immunogenic to the body's immune system. Cancer cell membrane-derived nanoparticles antigenically recapitulate the parental cancer cells and can be exploited to induce immune response reactive with tumor-associated antigens (TAAs). The use of the cancer cell membrane-derived nanoparticles to deliver immunostimulatory adjuvants facilitates the cross-presentation of tumor antigens by antigen-presenting cells and their costimulation, triggering potent antigen-specific T responses to eliminate established tumors. These nanoparticles can be engineered to carry immunostimulatory signals to facilitate the cross-presentation of TAAs and the induction of potent antitumor immunity. In this study, cancer cell membrane-based vesicles (CCMVs) are prepared from B16 melanoma cells and engineered to deliver the immunological agent polyinosinic:polycytidylic acid (poly-IC). We show that CCMV is preferentially uptaken by bone marrow-derived dendritic cells (BMDCs) as compared to other cell types (macrophages, fibroblasts). The efficient delivery of poly-IC to BMDCs results in enhanced antigen cross-presenting capability of BMDCs and T-cell activation. Additionally, immunization of mice with poly-IC-carrying CCMV elicits a potent antitumor immune response. In conclusion, poly-IC-decorated tumor-derived CCMV may be used as a therapeutic vaccine to potentiate antitumor immunity.
1 INTRODUCTION
Tumor-associated antigens (TAAs) can be utilized to develop immunotherapeutics, such as cancer vaccines.1, 2 Strategies that introduce TAAs into antigen-presenting cells (APCs), e.g., macrophage and dendritic cells, for efficient cross-presentation and optimized T-cell priming are critically required to generate antigen-specific T-cell immunity. Cancer cell membrane offers distinctive advantages over the other sources of TAAs (e.g., tumor lysates) for immunotherapeutics development.3-5 It not only recapitulates, at least partially, the antigenic repertoire of source cancer cells but also significantly enriches TAAs. Both autologous whole tumor cells and tumor cell lysates have been investigated as sources of TAAs for tumor vaccination in both preclinical studies and clinical trials in the past two decades.6, 7 However, the insufficient uptake by APCs, abundant in lymph nodes, leads to low antigenicity and low immune activation, hence limiting the use of whole tumor cells and tumor cell lysates as cancer vaccines.7 Recently, cancer cell membrane-coated nanoparticles alone or loaded with an immunostimulatory adjuvant have been studied to facilitate antigen-specific antitumor immune responses.4, 8 Cancer cell membrane-coated nanoparticles emerge as a promising platform for achieving tumor-specific delivery of therapeutics.9, 10
Several approaches have been reported for the preparation of cancer cell membrane-derived nanoparticles. For instance, cancer cell membrane can be directly coated onto the surface of polymer-based nanoparticles, such as poly(lactic-co-glycolic acid) (PLGA) nanoparticles, to present a full array of cancer cell membrane antigens. Immunostimulatory adjuvant or agents can also be loaded into the nanoparticle core to boost tumor-specific immunity.4, 11 Zhang and coworkers collected cell membrane from breast cancer cells and coated them onto biodegradable PLGA nanoparticles, which were further coupled with TLR-4 activator as an immunological adjuvant. The resulting nanostructures were shown to efficiently deliver TAAs to APCs and subsequently induce specific T-cell activation.11 Later on, his group loaded CpG oligodeoxynucleotide 1826 into PLGA nanoparticles and coated PLGA nanoparticles with cancer cell membrane to promote immunity against multiple tumor antigens.4 To increase the specific uptake of cancer cell membrane-coated nanoparticles by APCs, Liu and coworkers constructed a nanovaccine formulation by coating adjuvant-containing PLGA nanoparticles with mannose-modified cancer cell membranes for active cancer immunotherapy. The obtained nanovaccine showed enhanced uptake by APCs, triggering more potent antitumor immune responses.3 Despite these impressive outcomes derived from the above studies, particular challenges exist for the clinical use of cancer cell membrane-coated nanoparticles. The release of the encapsulated immunostimulants depends on the degradation of the nanoparticle core, potentially causing a delayed immune response. Additional manufacturing considerations have to be taken into account on the use of exogenous nanoparticulate materials in immunotherapeutic development, such as scale-up, product consistency, and translational hurdles. Alternatively, the lack of exogenous material use (like nanoparticle core) makes cancer cell membrane-derived nanovesicles promising platforms for autologous cancer cell vaccination. PEGylated cancer cell membrane-derived nanovesicles loaded with CpG and PEGylated nanovesicles exhibited efficient drainage to local lymph nodes.12 Lim and coworkers designed cancer cell membrane-derived nanovesicles (termed as tumosomes) by reconstituting cancer cell membranes containing exogenous helper lipids (cholesterol and synthetic phospholipids) and two kinds of lipid-based immunomodulatory adjuvants. The resulting tumosomes showed enhanced antitumor efficacy and extended the total survival rate of the treated animals.13 The Mooney group reported a relatively simple form of cancer cell membrane-derived nanovesicle. They observed that cancer cells could be disrupted vigorously to form membrane-enclosed vesicles before the loading of immunostimulants. These structures have been shown to facilitate the induction of antigen-specific cellular and humoral immune responses in animal models.14 This nanoparticle-based cancer vaccine was relatively large (~500 nm) with a heterogeneous size distribution. It has been widely recognized that tumor-draining lymph nodes are the primary site for priming of TAA-specific T cells by DCs.15 Large particle size may be suboptimal as it can impede the passive drainage of nanoparticle-based cancer vaccine to the lymph nodes.16, 17
In this work, we developed cancer cell membrane-based vesicles (CCMVs) with significantly reduced size (~120 nm) and uniform size distribution (polydispersity index [PDI] < 0.2). We chose B16 mouse melanoma cells as a source and prepared CCMV loaded with the polyinosinic:polycytidylic acid (poly-IC), a highly immunostimulatory agent. Poly I:C, a synthetic analog of double-stranded RNA (dsRNA), is recognized by toll-like receptor 3 to mimic pathogen infection and boost immune system activation to promote anticancer therapy. Recently, one modified form of poly-IC (Hiltonol®) is being evaluated in clinical trials as single agents and in combination with other immunotherapeutic drugs. We assessed the feasibility of using CCMV to deliver immunostimulatory agents and enhance the immune responses against cancer (Figure 1). Our results show that poly-IC CCMV promotes antigen presentation and T-cell activation by preferentially targeting the activation of APCs. Administration of poly-IC CCMV to mice leads to markedly stronger inhibition of melanoma progression as compared with free poly-IC.

2 RESULTS
2.1 Characterization of CCMV
The maximum loading of poly-IC in the CCMV was achieved when the mass ratio of poly-IC to membrane-associated proteins was kept at 1:50 (w/w) (Figure 2A). Dynamic light scattering (DLS) data showed that the final size of poly-IC CCMV was approximately 120.5 ± 12.4 nm and its zeta potential was −25.5 ± 3.8 mV (Figure 2B). After 1 week of storage at 4°C, the mean size of poly-IC CCMV did not changed significantly (124.6 ± 15.7 nm), suggesting the good colloidal stability of poly-IC CCMV. To preserve the bioactivity of TAAs and poly-IC, poly-IC CCMV used in this work was freshly prepared and used within 1 week. The protein profile of the extracted CCMV was examined using Western blot analysis (Figure 2C). Compared with the whole-cell lysate, plasma membrane biomarker (Na+/K+ ATPase α-1) and tumor-associated antigens Gp100 and GRP170 were enriched in CCMV (Figure 2D), similar to our previous observation.18 Notably, compared to the enriched plasma membrane marker Na+/K+ ATPase α-1 in the cell membrane fraction, both GRP170 in the endoplasmic reticulum and Gp100 expressed in melanosomes decreased. This result suggests the predominant content of CCMV is from the plasma membrane. The transmission electron microscopy image of poly-IC CCMV reveals a vesicle-like structure with a size range similar to that measured by DLS (Figure 2E).

2.2 CCMV facilitates the internalization of poly-IC by bone marrow-derived dendritic cells (BMDCs)
Compared with vesicles derived from the plasma membrane of macrophages or fibroblasts, DiD-labeled CCMV (DiD-CCMV) showed the highest accumulation in BMDCs (Figure 3). To evaluate if CCMV can facilitate the entry of poly-IC into BMDCs, we imaged and quantified the cellular uptake of fluorescein isothiocyanate (FITC)-labeled poly-IC, which was either in free form or complexed with CCMV. Upon complexing with CCMV, the internalization of poly-IC by BMDCs was enhanced as compared with that of poly-IC in a free form (Figure 4A) and flow cytometry (Figure 4B). Therefore, tumor cell membrane-derived vesicles have been shown to improve the delivery of immunostimulants into APCs.


2.3 Poly-IC CCMV-treated BMDCs show the enhanced capability to prime tumor antigen-specific T cells
To test whether the increased delivery of poly-IC into APCs can result in their enhanced activation, we examined the levels of proinflammatory cytokines (i.e., interleukin-6 (IL-6) and IL-12p40) as well as the level of the costimulatory molecule CD86. CCMV alone brought about a 9-fold increase in IL-6 production and a 2-fold increase in IL-12p40 production. While CCMV mixed with poly-IC slightly further increased the levels of the cytokines, poly-IC CCMV induced the highest levels of IL-6 and IL-12p40 production, 20- and 3-fold higher than the control group, respectively (Figure 5A). In addition, the upregulation of the costimulatory marker CD86 on DCs (right shift) was observed in the poly-IC CCMV treatment group (Figure 5B). To determine the impact of poly-IC complexed with CCMV on the capability of BMDCs to cross-present TAA, we cocultured BMDCs treated with the indicated formulations and Gp100-specific Pmel cells. Consistent with the strongest BMDC activation induced by poly-IC CCMV, we found that the BMDCs treated with poly-IC CCMV were most effective in stimulating Pmel cells, as indicated by a 3-fold increase in IL-2 production (Figure 5C) and a 2.4-fold increase in the frequency of IFN-γ-producing cells (Figure 5D). The results suggest that poly-IC CCMV efficiently delivers TAA (e.g., Gp100) and costimulation signals to APCs for robust activation of antigen-specific T cells.

2.4 Poly-IC-loaded CCMV displays potent therapeutic efficacy in vivo
To assess if the enhanced antigen cross-presentation by poly-IC CCMV treatment can translate into immune-mediated control of tumor in vivo, we treated B16 tumor-bearing mice with poly-IC CCMV or other formulations. Although CCMV alone or CCMV mixed with free poly-IC showed modest inhibitory effects on tumor growth, poly-IC CCMV was the most potent in delaying tumor growth, causing a 4-fold reduction in tumor volume as compared to the control group (Figure 6A). Consistent with tumor inhibition, vaccination with poly-IC CCMV resulted in highly elevated CD86 expression on tumor-infiltrating DCs as well as a marked increase in the frequency of CD8+ or CD4+ T cells that express IFN-γ, as determined by intracellular cytokine staining and flow cytometric analysis (Figure 6B). Transcription of IFN-γ (160-fold higher) and IL-12p40 (20-fold higher) was also significantly enhanced in the tumors treated with poly-IC CCMV compared to the untreated group (Figure 6C). These results indicate that CCMV represents an efficient platform to deliver TAA and immunostimulatory agents for cancer immunotherapy.

3 DISCUSSION
Cancer cell membrane-based vesicles retain a unique antigen repertoire that represents an antigenic fingerprint of parental cancer cells. In this work, CCMVs are fabricated using mouse B16 melanoma cells and loaded with the immunostimulatory poly-IC. The in vitro studies of APC activation, T-cell priming, and in vivo therapeutic evaluation have demonstrated that CCMV engineered to carry immunostimulatory signals can efficiently enhance DC activation and antigen cross-presentation to induce cellular antitumor immunity.
The obtained CCMV possesses a nanoscale structure with a negative surface charge (~−25 mV) that is similar to the source B16 tumor cells. The western blot analysis reveals that the typical plasma membrane biomarker Na+/K+ ATPase α-1 is enriched in CCMV. However, the levels of the melanoma antigen gp100 that resizes in premelanosome19 and endoplasmic reticulum-resident protein GRP17020 are low. The nanoscale size (~120 nm) of the prepared CCMV also makes them more readily accessible by APCs in vivo.16, 17
Cancer immunotherapy harnesses the host immune system to eliminate cancer cells, and it provides an innovative strategy for the treatment of various cancers, including melanoma.21 A therapeutic cancer vaccine, one form of cancer immunotherapies, is designed to mobilize a T-cell-mediated immune response against tumor-specific antigen or TAA. Upon activation, immune effector cells such as antigen-specific T cells will hunt and attack cancer cells that present these antigens on the cell surface.22, 23 The insufficient generation and expansion of functional cytotoxic T lymphocytes (CTLs) in the tumor microenvironment (TME) represent a major hurdle for successful cancer immunotherapies, including immune checkpoint blockade. Studies have shown that rationally incorporating immunostimulatory agonists into cancer immunotherapy or the use of these agents in combination with nanoparticle and microparticle-based therapeutic platforms can synergistically augment immune responses at a relatively low dose while reducing potential toxicity.24-27 To induce T-cell activation more efficiently, we strategically incorporate the widely used immunostimulator poly-IC into the nanoscale CCMV vaccine. Our data show that melanoma cell-derived vesicles, that is, CCMV, are more susceptible to internalization by DCs as compared with vesicles derived from either macrophages or fibroblasts. This enhanced internalization results in a more efficient entry of poly-IC into DCs upon complexing with CCMV, a necessary step for DC activation. Compared with free poly-IC, CCMV complexed with poly-IC induces markedly enhanced DC activation, as evidenced by increased production of cytokines and expression of the costimulatory molecule, both of which are essential for T-cell priming. Indeed, poy-IC CCMV treated DCs acquires a superior capacity in driving antigen-specific T-cell activation and proliferation. Therefore, our novel vaccine formulation has several advantages, nanoscale vesicle size, the presence of TAA reservoir in the vesicles, and their capability of activating DCs and providing immune costimulation. Importantly, the incorporation of poly-IC into the CCMV vaccine platform generates a significantly improved antitumor immunity in vivo, as evidenced by potent tumor growth inhibition and immune activation at the tumor sites.
During the progression of solid tumors, cancer cells orchestrate an immunosuppressive TME.28, 29 This suppressive TME supports tumor progression by inducing tumor escape from recognition and destruction by CTLs or compromising tumor response to cancer therapies, including cancer immunotherapies.30 Among several mechanisms, APC inertness in the TME is attributed to T-cell dysfunction.31, 32 Our data demonstrate that poly-IC CCMV causes significant immune changes in the TME, including activation of DCs and recruitment of CD4 and CD8 T lymphocytes. Thus, our engineered poly-IC CCMV platform may be exploited to convert the immunosuppressive TME to a highly immunostimulatory one and further enhance tumor response to other immunotherapies, such as immune checkpoint inhibitors.
In summary, CCMV is developed as a platform to deliver autologous TAAs and costimulatory signals (e.g., poly-IC) to APCs for antigen cross-presentation and T cell activation. Personalized immunotherapy may be developed on the basis of this biomimicry approach.
4 MATERIALS AND METHODS
4.1 Materials
Vybrant™ Multicolor Cell-Labeling Kit (DiO, DiI, DiD Solutions) was purchased from Thermo Scientific. Mini extruder and polycarbonate porous membranes with different pore sizes were purchased from Avanti Polar Lipids. Dulbecco's modified Eagle's medium (DMEM), penicillin–streptomycin (10,000 U/ml), and trypsin-EDTA (0.25%) were obtained from Life Technologies. Unless noted, all other chemicals were purchased from Sigma-Aldrich. Antibodies against Na+/K+ ATPase α-1 (clone C464.6; Millipore) and Pmel17 (sc-377325; Santa Cruz) were used in western blot analysis. Poly-IC- and FITC-labeled poly-IC were purchased from Invivogen. Fluorescently labeled mouse monoclonal antibodies to CD11c (HL3), CD86 (GL-1), CD4 (GK1.5), CD8a (53-6.7), CD3 (HL3), IFN-γ (XMG1.2), IgG1 (RTK2071), and isotype control rat IgG2b (RTK4530) were obtained from BioLegend. ELISA (enzyme-linked immunosorbent assay) Kits for mouse IL-6, IL-12p40, and IL-2 were purchased from BioLegend.
4.2 Mice and cell lines
C57BL/6N mice were purchased from Envigo. Pmel transgenic mice carrying TCR transgene specific for the mouse homolog (pmel-17) of human gp100 were obtained from the Jackson Laboratory. B16 melanoma cell line, NIH3T3 cells, and J774A.1 cells were cultured in DMEM media supplemented with 10% heat-inactivated fetal bovine serum (Life Technologies), 2 mM l-glutamine, 100 U/ml penicillin, and 100 mg/ml streptomycin. All experimental animal procedures were conducted under the protocols approved by the Institutional Animal Care and Use Committee of Virginia Commonwealth University.
4.3 B16 cell membrane extraction and characterization
To prepare one batch, 2.5 × 108 B16 cells were suspended in 4 ml of homogenization buffer (225 mM mannitol, 75 mM sucrose, 30 mM Tris-HCl, 1 mM MgCl2, 1 mM KCl, 2 mM phenylmethylsulfonyl fluoride, EDTA-free protease inhibitor, RNase 10 µg/ml, and DNase 10 μg/ml, pH 7.4). The cells were gently homogenized on ice using a Potter–Elvehjem homogenizer (1500 rpm and 20–30 passes) and centrifuged at 500g for 10 min. The supernatant containing the cell membrane fractions was collected. The pellets containing intact cells were subjected to extra rounds of homogenization and centrifugation as needed until most cells were free of intact membrane structure observed under the light microscope. All the supernatants were pooled and centrifuged at 10,000g for 30 min. The pellet containing the mitochondrial contamination was discarded, and the supernatant was further centrifuged at 25,000g for 20 min to collect the pellet. The pellet containing crude cell membrane was suspended in resuspension buffer (5 mM Bis-Tris and 0.2 mM EDTA, pH 6.0) and layered on top of an 11-ml discontinuous sucrose gradient, composed of 55%, 43%, and 38% (w/v) sucrose in the resuspension buffer. The discontinuous gradients were centrifuged in a Sorvall™ TH-641 Swinging Bucket Rotor at 95,000g for 2 h. The plasma membrane-rich region (interface between 43/55%) was collected, diluted 10-fold with resuspension buffer, and centrifuged at 95,000g for 1 h to form pure cell membrane pellet. All centrifugations were performed at 4°C. The collected cell membrane was resuspended in saline solution and stored at −80°C for future use.
Western blot analysis was performed to examine whether the extracted cell membranes could recapitulate plasma membrane biomarker (Na+/K+ ATPase α-1) and those known B16 membrane-associated proteins, including glucose-regulated protein 170 (GRP170) and glycoprotein 100 (Gp100).
Extracted cell membranes were further analyzed in terms of quantities of membrane-associated proteins and membrane-associated phospholipids. Membrane-associated proteins were quantified using a Pierce™ BCA Protein Assay Kit (Pierce Chemicals), and membrane-associated phospholipids were quantified using a Phospholipid C Assay Kit (Wako Pure Chemical Industries, Ltd.). One batch of purified B16 membrane fractions (from 2.5 × 108 cells) typically contained an approximate total of 0.86 mg proteins and 0.42 mg phospholipids. The extracted cell membranes were stored at −80°C and were used within 1 month.
4.4 Preparation and characterization of poly-IC-encapsulating CCMV
Poly-IC was dissolved in phosphate-buffered saline (PBS) at the concentration of 1 mg/ml. Various amounts of poly-IC were mixed with B16 cell membrane on the basis of 5 mg of membrane-associated proteins at 1:200, 1:100, 1:50, and 1:20 (w/w), sonicated (Qsonica Q700 equipped with 3.2 mm stepped microtip probe) for 10 s at the 60% amplitude and then extruded through 1000, 400, and 200 nm polycarbonate membranes in sequence. Following centrifugation at 21,000g for 1 h, the pellet of poly-IC CCMV was collected and suspended in 0.5 ml of saline containing 5% sucrose. The supernatant was analyzed with a Quant-iT™ RNA Assay Kit (Invitrogen) for the quantification of free poly-IC according to the manufacturer's instructions. Poly-IC loaded in each formulation was reported as pmol poly-IC/mg membrane-associated proteins. The size and surface zeta potential of poly-IC CCMV was determined via DLS measurements using a Malvern Zetasizer. Poly-IC CCMV formulations were imaged under a JEM-3010 transmission electron microscope.
4.5 Assessment of vesicles made of different cell membranes by APCs
BMDCs were generated through the culture of mouse BM cells in the presence of granulocyte–macrophage colony-stimulating factor as described previously.33 To examine uptake by BMDCs, leukocytes (J774A.1), fibroblast cells (NIH3T3), or B16 tumor cells were labeled with DiD according to the supplier's recommended protocol before cell plasma membrane collection. The DiD-labeled cell membranes were constructed into nanovesicle as described above and added to the BMDCs (each type of cell membrane vesicle was applied under the same amount of cell membrane-associated proteins, 0.5 µg/ml) and incubated for 2 h. The cells were rinsed with cold PBS three times and subjected to fluorescence imaging.
4.6 Evaluation of delivery of poly-IC to APCs by CCMV
BMDCs were incubated with CCMV that were physically mixed or complexed with FITC-poly-IC (2 µg/ml) at 37°C for 15 min. FITC fluorescence-labeled poly-IC was kept at an equivalent concentration for comparison. The cells were rinsed with warm PBS three times and detected under a fluorescent microscope and flow cytometry. In a separate experiment, BMDCs were incubated in the presence of empty CCMV and a mixture of CCMV and poly-IC, poly-IC CCMV, or left untreated (control) for 24 h. Protein levels of IL-6 and IL-12p40 or CD86 expression in the culture media were determined by ELISA and flow cytometry, respectively.
4.7 In vitro priming of antigen-specific T cell
BMDCs were incubated with CCMV and a mixture of CCMV and poly-IC, poly-IC CCMV, or left untreated for 5 h. BMDCs (1 × 104) were then cocultured with purified Pmel CD8+ T cells (1 × 105) for 3 days. The IL-2 level in the culture media was determined using an ELISA Kit. For staining intracellular cytokines, Pmel cells primed by BMDCs were treated with phorbol 12-myristate 13-acetate (50 ng/ml), ionomycin (400 ng/ml), and brefeldin A (5 μg/ml) for 5 h. The cells were further stained with anti-CD8 antibody, fixed, and permeabilized using a Cytofix/Cytoperm Kit (BD Biosciences). The cells were stained with an anti-mouse IFN-γ antibody.
4.8 Tumor studies
C57BL/6N mice were inoculated with 2 × 105 B16 cells on the right flank on Day 0. Three days after tumor inoculation, the mice were intravenously administered with blank CCMV (40 mg of membrane-associated proteins/kg) and a mixture of CCMV and poly-IC, poly-IC-CCMV for three doses (0.8 mg of poly-IC/kg) at 3-day intervals or left untreated. Tumor growth was monitored. The tumor size was measured and tumor volume was calculated every other day based on the following formula = width2 × length × 0.5. For analysis of immune cell infiltration, tumors were harvested, minced, and digested with collagenase type II (1 mg/ml) and DNase I (100 μg/ml) at 37°C for 2 h. Single-cell suspensions were obtained by filtering through a 70-μm cell strainer, followed by Histopaque (Sigma-Aldrich) gradient centrifugation to isolate viable cells. Tumor-derived single cells were treated with phorbol myristate acetate (10 nM) and ionomycin (0.5 μM) for 6 h. Brefeldin A (5 μg/ml) was then added in the last 3 h of culture. Cells were stained with antibodies to CD11c, CD86, CD8a, CD4, and CD3, followed by fixation, permeabilization and intracellular staining of IFN-γ.
4.9 Real-time PCR
Total RNA was extracted from the tumor tissue using TRizol reagent according to the manufacturer's protocol (Invitrogen) and reverse transcripted with Superscript II reverse transcriptase. Real-time PCR was detected on the ABI 7900HT Fast Real-time PCR System using TaqMan® Universal PCR Master Mix and TaqMan® Gene Expression Assays probe (Applied Biosystems). The Assay Identification number of ifng is Mm01168134_m1, and il12p40 is Mm01288989_m1. Amplification reactions for each sample were tested in triplicate, and the results were normalized to the ACTB gene expression level as an internal reference. The fold change in studied gene expression was calculated using the 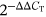 method.
method.
4.10 Statistical analysis
The data are expressed as mean ± SD. The statistical analysis was performed using one-way analysis of variance, and Student's t-test was conducted for pairwise comparison of subgroups. The results were considered statistically significant at a p < 0.05 in all cases.
AUTHOR CONTRIBUTIONS
Hongliang He: Investigation (equal); methodology (equal); writing—original draft (equal). Chunqing Guo: Investigation (equal); methodology (equal); writing—original draft (equal). Wenjie Liu: Methodology (supporting). Shixian Chen: Methodology (supporting). Xiang-Yang Wang: Conceptualization (equal); project administration (equal); writing—original draft (supporting); writing—review and editing (equal). Hu Yang: Conceptualization (equal); project administration (equal); writing—original draft (supporting); writing—review & editing (equal). All authors have read and approved the final manuscript.
ACKNOWLEDGMENTS
The authors thank Nanomaterials Core Characterization Facility of Virginia Commonwealth University for particle imaging.
CONFLICT OF INTEREST
Hu Yang is an Associate Editor of MedComm - Biomaterials and Applications. The author was not involved in the journal's review or decisions related to this manuscript. The remaining authors declare no conflict of interest.
ETHICS STATEMENT
The animal procedures were approved by the Institutional Animal Care and Use Committee of Virginia Commonwealth University (approval number AD20158).
Open Research
DATA AVAILABILITY STATEMENT
All the data and materials are available in this manuscript.

