

Cooling rate and dilution affect the nanostructure and microstructure differently in model fats
Abstract
The effects of cooling rate and solid mass fraction on the polymorphism, nano and microstructure, thermal and rheological properties of binary mixtures of fully hydrogenated canola oil and canola oil at 20°C have been studied. The β-polymorph was observed in fully hydrogenated canola oil (FHCO) when crystallized at slow cooling rates (0.1C°/min), however crystallization at higher cooling rates (0.7 and 10°C/min) resulted in the formation of the α form. The β-polymorph was detected in all the binary mixtures of FHCO/canola oil and was not affected by crystallization at different cooling rates. Melting thermograms obtained from 100% FHCO displayed three melting peaks, associated with the development of the β-polymorph via α→ β′→ β-polymorphic transition in the DSC pan. Some solubilization of solid FHCO into canola oil was observed and the solubility was proportionally higher with increasing liquid oil fraction. The strong influence of the matrix concentration on micro/nanoscale structure was demonstrated by characterization of crystal size using cryogenic transmission electron (Cryo-TEM) and polarized light microscopy (PLM). Crystallization under higher cooling rates lead to formation of smaller nano and meso-structural elements. Furthermore, oscillatory rheology showed the influence of structural elements' size and polymorphism on material strength. The shear storage modulus (G′) of the mixtures was higher when crystallized at fast cooling rates (10°C/min). In contrast, for pure FHCO, G′ increased by lowering the cooling rate and the highest storage modulus was observed after crystallization at 0.1°C/min.
Abbreviations:
FHCO, fully hydrogenated canola oil; Cryo-TEM, cryogenic transmission electron microscopy; PLM, polarized light microscopy; SFC, solid fat content
Introduction
The physical and functional properties of fats are largely determined by the three-dimensional organization of a fat crystalline network and the polymorphic state of the solid TAGs 1-4. Formation of the fat crystal network, crystalline morphology and phase behavior are modified by factors such as TAG composition and fatty acid positional distribution, as well as crystallization conditions including cooling rate or crystallization temperature, agitation, storage time and so on 5. Most fats and oils have limited applications due to their particular chemical compositions, in particular fully hydrogenated vegetable oils. However, these same fully hydrogenated oils are readily available, locally grown, relatively inexpensive, and contain very low levels of trans fatty acids. Therefore, to extend the use of these hydrogenated stocks, they can be modified either chemically by interesterification or physically by blending with liquid vegetable oils combined with crystallization under specific conditions (cooling and shear rates).
Several authors have investigated the effects of cooling and shear rates, as well as blending ratios, on the crystallization behavior of non-interesterified mixtures of oils with high-saturate or fully hydrogenated fats 6-10. This work demonstrated the existence of different microstructures when the materials were crystallized under the influence of specific external fields. Some authors have reported that when a fat sample was cooled rapidly from the melt, the system crystallizes before reaching an equilibrium molecular packing due to mass transfer limitations. On the other hand, during a slow cooling process, the rate of viscosity increase is lower and the induction times of crystallization (the time involved achieving nucleation) are longer; therefore mass transfer limitations are minimized. This enhances the attainment of equilibrium organization of the molecules within the material 3, 11, 12. Some other studies introduced physical blending as another alternative to enhance the functional properties of crystallized fat materials 7. Blending with vegetable oils will decrease melt viscosity and the rate of crystallization. Nevertheless, it is important to understand the influence of blending on the structure of the forming system during crystallization and its relation to processing, so as to attain specific physical and functional properties in a fat.
The aim of this work was to explore the possibility of using fats with high contents of fully hydrogenated stocks such as, stearic acid, to tailor functional properties and nutritional benefits of these hard fats with minimal processing and without resorting to interesterification. Here we report on the effects of blending and cooling rates on the structure, at different length scales, of fully hydrogenated canola oil (FHCO) and canola oil mixtures (in 10% increment from 50% to 100% (w/w) of hardstock and consequently its effects on the rheological properties of the systems.
Materials and methods
Materials
FHCO was generously provided by Bunge Canada (Toronto, Canada). Canola oil was purchased from local supermarkets. FHCO contained approximately 88% stearic acid (18:0), 9% of palmitic acid (16:0), and 2% arachidic acid (20:0).
Samples preparation and treatments
FHCO was diluted with canola oil in 10% increments from 50% to 100% (w/w).
The mixtures were heated to 100°C in an oven and held at this temperature for 10 min to erase the crystal memory. All the blends of canola oil and FHCO were crystallized from 80°C to 20°C at two different cooling rates; 10 and 0.7°C/min, and the pure FHCO was also crystallized at 10, 0.7 and 0.1°C/min. The molten blends were then poured into aluminum cylindrical molds (20 mm diameter and 3.2 mm height) connected to a temperature controlled water bath (Neslab RTE-111, Fisher Scientific, St. Louis, MO) and the cooling capabilities of the water bath was used for the two lowest cooling rates. The highest cooling rate was achieved by quenching the samples with liquid nitrogen. The 80°C molten physical mixtures were transferred into the precooled molds and immediately cooled down with liquid nitrogen. Blends were subsequently stored for 1 day in an incubator set at 20°C before rheological measurements. Additionally, a small piece of this fat disc was employed for the cryogenic transmission electron microscopy (Cryo-TEM) studies.
X-ray analysis
The polymorphic modifications of crystallized samples were determined by powder X-ray diffraction (XRD). A Multiplex Powder X-ray diffractometer (Rigaku, Japan) with a 1/2 degree divergence slit, 1/2 degree scatter slit, and a 0.3 mm receiving slit, was set to 40 kV and 44 mA and used to analyze samples polymorphism. Approximately one gr of the sample was placed into a pre heated glass x-ray slide. After 24 h of storage in an incubator set to the appropriate crystallization temperature scans were performed from 0° to 30° at 2°/min. Results were analyzed by using MDI's Jade 6.5 software (Rigaku, Japan). Three determinations of five replicates were performed.
Rheology measurements
A TA Instrument AR2000 controlled stress dynamic Rheometer (TA Instruments, Mississauga, ON, Canada) was used to perform the rheological measurements. Oscillatory tests (small deformation) were performed by means of a strain sweep experiment at a frequency of 1 Hz, within the range of 1 × 10−3 to 0.8% strain, and maximum applied normal force of 5N. The experiments were carried out using a stainless steel parallel plate (20 mm diameter); the normal force was set approximately at 10 N for all disc samples. The sample platform temperature was controlled, allowing for sample temperature to be maintained at 20°C during analysis. To prevent slippage, sandpaper (grade 60) was attached to the lower surface of the geometry and the upper surface of the Peltier base of the rheometer. The reported data are the average of 8–10 individual replications.
Solid fat content (SFC) measurements
Melted samples were poured into preheated NMR glass tubes and stored for 24 h at the crystallization temperature to perform the SFC measurements. SFC was measured by pulse NMR (p-NMR) using a Bruker Minispec spectrometer, (Bruker Optics Ltd., Milton, ON, Canada). The reported data corresponds to the average of three individual measurements.
DSC measurements
The thermal behavior of the samples was studied by DSC, DSC, (TA Instrument, Q1000, New Castle, DE, USA). 5–10 mg of the samples were placed in DSC pans and were subjected to the following temperature program in the DSC cell: heating at 100°C for 10 min to erase all the crystal memory, cooling at a rate of 10 0.7, and 0.1°C/min to the crystallization temperature. Having enough SFC to obtain the melting point, the cell was stabilized at the final crystallization temperature (100°C)for 60 min. Sample pans were stored in an incubator set at 20°C for 24 h before melting. Then, to obtain the peak melting temperature blends were heated at a rate of 1°C/min until 80°C. Samples were run in triplicate.
Thermograms were evaluated using TA Instruments Universal Analysis Software (TA Instruments – Waters LLC, New Castle, DE, USA). The onset and the peak melting temperatures (or simply melting point) of the samples were obtained from the thermograms and are shown in Table 1. Peak melting temperatures are related to the temperature at which the largest proportion of solid species in the fat sample melt, and are considered an average of the melting point of the sample. Three replicates were performed for each sample.
| SFC (%) | Melting point (°C) | |||||
|---|---|---|---|---|---|---|
| FHCO (%) | Fast cooling | Slow cooling | Fast cooling | Slow cooling | ||
| To | Tb | To | Tb | |||
| 50 | 39.5 ± 0.1 | 39.3 ± 0.1 | 42.62 ± 0.3 | 65.35 ± 0.3 | 42.83 ± 0.1 | 65.95 ± 0.1 |
| 60 | 50.1 ± 0.2 | 49.5 ± 0.2 | 42.88 ± 0.4 | 66.28 ± 0.4 | 43.19 ± 0.1 | 66.87 ± 0.1 |
| 70 | 61.5 ± 0.1 | 62.0 ± 0.1 | 43.12 ± 0.3 | 67.55 ± 0.7 | 43.31 ± 0.2 | 67.97 ± 0.2 |
| 80 | 73.7 ± 0.2 | 73.6 ± 0.1 | 47.08 ± 0.2 | 68.0 ± 0.24 | 47.38 ± 0.2 | 67.3 ± 0.04 |
| 90 | 86.2 ± 0.1 | 86.8 ± 0.1 | 51.86 ± 0.2 | 68.52 ± 0.2 | 51.19 ± 0.1 | 69.01 ± 0.1 |
| 100 | 98.8 ± 0.0 | 98.4 ± 0.1 | 57.33 ± 0.2 | 69.7 ± 0.3 | 57.47 ± 0.1 | 68.94 ± 0.3 |
 ((1))
((1))Polarized light microscopy (PLM)
PLM was used to observe fat microstructure. A small drop of preheated fat blend (80°C) was placed on a preheated slide at the same temperature, and a cover slip was then gently laid over the fat drop to remove air and spread the fat. Samples were transferred into a thermostatically controlled microscope stage (Model LTS 350, Linkam Scientific Instruments, Surrey, UK) to crystallize the fat blends at the above-mentioned cooling rates. Samples were imaged and processed using a Leica DM RXA2 microscope with polarized light (Leica Microsystems, Richmond Hill, Canada) and equipped with a CCD camera (Q Imaging Retiga 1300, Burnaby, BC, Canada). All images were acquired using a 40X objective lens (Leica, Germany). The camera was set for autoexposure. Openlab 5.5.0 software (Improvision, Waltham, MA, USA) was used to acquire images. Focused images were stored as uncompressed 8-bit (256 grays) grayscale TIFF files with a 1280 × 1.024 spatial resolution. Five images were captured from each of five replicates prepared.
Image analysis was carried out by Adobe Photoshop CS5 software (Adobe Systems Inc., San Jose, California, USA) and filters from the Fovea Pro 4.0 software (Reindeer Graphics, Inc., Asheville, NC, USA). A manual thresholding was applied to all the pictures to convert the grayscale images to binary images, in order to discriminate between features and background and to measure the features sizes. The microstructural elements were determined using the filter tools included in the Fovea Pro software.
Cryogenic transmission electron microscopy (Cryo-TEM)
A solvent washing technique was used to study the nanostructure of crystallized fat blends using Cryo-TEM 12-14. To discard oil fraction and favor single crystals observation in the fat blends, samples (crystallized at determined conditions) were dispersed in cold isobutanol (10°C) in fat/solvent ratios ranging from 1:25 to 1:50 according to the amount of oil present in the sample. The fat plus isobutanol mixtures were homogenized at 30,000 rpm with a rotostator (Power Gen 125, Fisher Scientific) for ∼10 min. Then, the crystals were collected by vacuum filtration through a glass fiber filter of 1.0 µm pore size. After filtration, the recovered solid was resuspended in cold isobutanol and rehomogenized to obtain a suitable dispersion of crystals. Finally, the mixtures were sonicated at 10°C for 60 min using an ultrasonic processor (Bransonic 1210R-DTH, Branson Ultrasonic Corporation, Danburry, CT, USA) to aid crystals dispersion. Five microliters of dispersion were placed on a copper grid with perforated carbon film (Canemco-Marivac, Quebec, Canada), and excess liquid was blotted using filter paper. A staining aqueous solution of 2% of uranyl acetate was used to enhance contrast. Subsequently, the sample was transferred to a cryo-holder (Gatan Inc., Pleasanton, CA, USA) for direct observation at −176°C in a FEI Tecnai G2 F20 Cryo-TEM (Eindhoven, The Netherlands) operated at 200 kV in low dose mode. Images were taken using a Gatan 4k CCD camera. Micrographs were stored and analyzed using DigitalMicrograph software (USA). Image J 1.42q software (USA) was employed for a semiautomatic analysis procedure.
Box-counting fractal dimension
The crystallized samples fractal dimension (Db) was determined by image analysis of the PLM images 15, 16. The Adobe Photoshop program™ was used for autothresholding of the images. After thresholding, images were input into Benoit 1.3 (TruSoft Int'l Inc., St. Petersburg, FL), a commercial available software, to calculate the 2D fractal dimension of the samples using box counting method. A grid formed by boxes of decreasing sizes is placed over the binary images and the number of occupied grids (N) is counted for a series of grid side length (L). The number of occupied boxes as a function of the size of the boxes is plotted and the slope of the log-log plot was determined as fractal dimension.
Statistical analysis
GraphPad Prism 5 software (GraphPad Software, Inc., CA, USA) was used to analyze the data obtained. Values reported correspond to the means and standard errors of the determinations. One-way ANOVA with a p-value <0.05 was used to evaluate statistical significance between the means.
Results and discussion
The polymorphic behavior of the mixtures of FHCO with canola oil was studied by powder XRD. The X-ray profile of the blends prepared in 10% increments of FHCO ranging from 50% to 100% (w/w) crystallized at 10°C/min are shown in Fig. 1A. Similar XRD patterns were observed for all blends cooled at 0.7°C/min, indicating that cooling rate does not affect significantly the samples' polymorphic behavior. Figure 1A clearly demonstrates that the crystal structure of FHCO was greatly influenced by the presence of canola oil. The existence of one strong diffraction peak in the WAXD region [wide-angle X-ray diffraction reflection] at 21.7 2θ (4.15 Å´) in 100% FHCO suggests the presence of the α-polymorphic phase in this sample. Dilution of FHCO with liquid oil induced changes in the positions of the diffraction peaks detected in the WAXD region. The observed diffraction peaks showed good agreement with the values reported for the β-form of FHCO; one sharp peak at 19.5 2θ (4.56 Å´) accompanied by at least two smaller peaks, one at 23.1 2θ (3.85 Å´) and the other at 24.1 2θ (3.69 Å´) 17, 18. This observation confirms the effect of liquid oil on the viscosity of the mixtures that enhances TAG diffusion and allows the adoption of more thermodynamically stable polymorphic forms 19-21. Similar results were obtained by analyzing the melting thermograms of the blends obtained from DSC profiles (Fig. 1B). Melting thermograms obtained from 100% FHCO displayed three melting peaks, associated with the development of the β-polymorph via α→ β′→ β polymorphic transition in the DSC pan. However, in the presence of canola oil, only a single sharp melting event in the range 63–69°C was observed in all the blends which suggests the existence of only the most stable polymorph, the β-phase. Analogous trends were observed by Ahmadi et al. for binary mixtures of FHCO and high oleic sunflower oil 16. Furthermore, Wright et al. found that canola oil dilution promotes the transformation from β′ to β form in anhydrous milk fat 21. As shown in Table 1, no significant differences were found between the samples' SFC and melting point due to the different applied cooling rates during crystallization. Campos et al. 22, on the other hand, reported a significant effect of cooling rate on melting profile and solid mass fraction of milk fat and lard. This discrepancy with our findings can be explained based on the large differences in TAG compositions between these systems and the range of SFC values studied.

(A) XRD patterns and (B) Melting profiles of different dilutions of FHCO and canola oil crystallized at 10°C/min at 20°C. (Similar behavior was observed for crystallization with cooling rates of 0.7°C/min).
A closer look at Fig. 1B, illustrates that increasing the canola oil mass fraction caused a gradual decrease in the melting temperatures of the blends. This finding may translate into differences in crystallization behavior and the resulting network properties of the samples. 16, 17. These changes observed in melting behavior can be explained by the depression of the freezing point (colligative effect) induced by the dilution of FHCO with canola oil. Colligative effects are governed by the number of solute particles existing in a particular volume of solvent, but not on the chemical properties of the solute 23. Thus, the decrease in the melting temperature of fat blends with increasing the amount of oil is the result of a higher TAGs solubility (Table 2). Comparable results were reported for the mixture of fully hydrogenated soybean oil in soybean oil (from 10% to 50% w/w) 7. Furthermore, these results can be also explained by differences in undercooling between blends. At any given temperature the more concentrated blends are exposed to a higher degree of undercooling than the more diluted samples and therefore had a greater driving force towards crystallization. On the other hand, no significant differences in melting behavior were observed by changing the cooling rate during crystallization. These findings suggest that the induced changes in supercooling did not affect further the samples' crystalline melting profile.
| FHCO (%) | SFC (%) measured | SFC (%) calculated | Tb measured | Tb calculated |
|---|---|---|---|---|
| 50 | 39.5 ± 0.1 | 51 ± 0.7 | 65.35 ± 0.33 | 62.95 ± 0.13 |
| 60 | 50.1 ± 0.1 | 58 ± 0.6 | 66.28 ± 0.41 | 64.50 ± 0.01 |
| 70 | 61.5 ± 0.1 | 65 ± 0.1 | 67.55 ± 0.79 | 65.8 ± 0.02 |
| 80 | 73.7 ± 0.2 | 76 ± 0.8 | 68.0 ± 0.24 | 67.3 ± 0.03 |
| 90 | 86.15 ± 0.2 | 84 ± 1 | 68.52 ± 0.28 | 68.02 ± 0.01 |
| 100 | 98.8 ± 0.2 | NC* | 69.7 ± 0.21 | NC* |
- * Not calculated.
Using pulse-NMR the SFC of the blends after 24 h of storage was determined and both the peak melting temperature, the average of the sample melting point, and the onset of melting of the blends were plotted as a function of SFC (Fig. 2A). This figure displays that the melting temperature of the blends increased linearly as a function of SFC. For better comparison both the peak melting temperature and the SFC of the blends are reported in Table 2. As expected, the amount of solid formed is dependent on the amount of FHCO and decreases with increasing the proportion of canola oil. Not surprisingly, the reduction in SFC was not proportional to the decrease in solids, expected based on dilution arguments. For instance, in the blends of 80% and 50% FHCO, the SFC was 74% and 39%, respectively. This reduction was due to solubilization of FHCO in the oil. At higher proportion of canola oil, more solids were solubilized and lower melting temperatures were observed.

(A) Changes in the onset (To) and peak melting temperature (Tb) of the FHCO blends as a function of SFC (B) Hildebrand plots for blends of FHCO and canola oil as a function of both calculated and actual SFC. (Data are related to the crystallization at 10°C/min and the results are comparable with those of 0.7°C/min).
The solubility of fat crystals in canola oil was predicted using Equation 1. In this study, the melting enthalpy of the 100% FHCO in the β-polymorphic (164.9 KJ/mol), and the molecular weight of FHCO (MW = 891.49 g/mol) were used to calculate the melting point and the SFC of the blends (reported in Table 2). As shown in this table, a deviation from ideal solubility was evident in all the blends where the calculated values for melting point and SFC differed from the measured values. Since canola oil contains a range of TAG species, the interaction between FHCO and these species may lead to the deviation from the ideal solubility 24, 25. One may notice that this deviation is smaller in blends with higher concentration of FHCO, in particular in those with SFC above 70%. These results are interesting since the deviation of TAG solubility can be also observed by DSC. Some solubilization of solids into canola oil was previously observed for milk fat, and the amount being proportionally higher with increasing dilution 21. The Hildebrand plot (Fig. 2B) shows a straight line with high correlation coefficients (r2) between the Ln versus 1/Tp for both measured and calculated SFC. This figure displays a higher SFC measured in the binary mixture of FHCO and canola oil than the calculated value; however, the measured and calculated SFC values are comparable at higher concentrations of FHCO (i.e., 90% FHCO). This finding may be explained by the differences in molecular volume between the solute and solvent at higher ratio of solvent-solute that influences the entropy of mixing 24. Knoester et al. 26 and Timms 27 reported deviations from ideal behavior when solid solutions or imperfect crystals with a greater solubility than perfect crystals are formed. Based on this theory, our results indicate the formation of perfect crystals of similar polymorphic forms and similar solid solution only at higher concentrations of FHCO in canola oil. Ahmadi et al. 16 reported a positive deviation of the predicted values of SFCs and melting points within the whole range of dilutions (from 10% to 90% of FHCO in Sunflower Oil). However, the discrepancy observed between these author's results and our findings may be attributed to differences in the nature of the samples as they differ in composition, and therefore in the enthalpy and melting point values.
To explore the effects of matrix dilution and different crystallization conditions on sample microstructure, PLM was used and the microstructure of the material at high FHCO concentrations was studied. PLM micrographs of fat blends crystallized at 0.7 and 10°C/min are presented in Fig. 3A–C and D–F, respectively. Even though only mixtures with proportions of 50, 90, and 100% FHCO are despicted, PLM images from blends within the whole range of hardstock proportion studied were obtained showing an analogous tendency to those observed in this figure. The crystalline morphology of all the blends, regardless the cooling rate used during crystallization,was predominantly spherulitic in nature, which indicates that matrix dilution with canola oil did not impact directly the primary organization of the mesoscale. Only a steady decrease in network density as a function of dilution, along with a spherulitic growth restricted by adjacent agglomerates in highly concentrated blends was evident (Fig. 3A–C and D–F). Analogous results are reported for comparable fat systems by other researchers 12, 16, 28, 29. The PLM microstructural observations can be explained by changes in the thermal behavior the resulted from increasing canola oil levels. Diluted blends had lower melting points (Table 2) resulting in a lower degree of undercooling during crystallization at a determined temperature. As a consequence, it is possible to observe in 50% FHCO systems a less compact crystalline network which suggests a decrease in the nucleation rate in the more diluted systems.

PLM images of FHCO/canola oil crystallized at: 0.7°C/min (A–C), and 10°C/min (D–F). Images belong to 50% FHCO (A and D), 90% FHCO (B and E), 100% FHCO (C and F).
These differences in the microstructural network density and thermal properties may also reflect the changes observed in sample polymorphism with dilution. As seen in Fig. 1A, an α-polymorphic form predominated in FHCO samples while only the β-polymorph was identified in the blends with canola oil.
To understand and therefore explain the performance of fats in terms of their nano-structural level, the nanostructure needs to be characterized. As explained in the Materials and Methods section, images of the nano-crystalline units of all the systems were obtained using Cryo-TEM. Examples of micrographs acquired for 50 and 100% FHCO crystallized at 0.7 and 10°C/min (presented in Fig. 4A–D respectively) demonstrate significant changes in the blends nano-platelet dimensions. Regardless of the cooling rates used during crystallization, nano-crystal size increased significantly with an increment of the liquid oil incorporated. These results were in close agreement with Acevedo and Marangoni findings for binary mixtures of FHCO and sunflower oil 12, 13. Nanoplatelet size in samples with 50% FHCO was approximately twice as large as that observed in 100% FHCO. This predominance of larger nano-sized particles in this diluted hardstock/oil binary mixture may be attributed to decreases in viscosity that promote TAG diffusion in the melt and allow the growth of the nano-crystals under lower supersaturation conditions. As expected, no changes in the platelets' morphology were induced by changes in FHCO concentration. It is important to note that different cooling rates affected the nanostructure of the blends more strongly under low supersaturation conditions. As previously explained, the ordering and packing of TAG chains into primary crystals is dependent of the viscosity of the melt. When a large change in supercooling conditions, given by an increase in the cooling rate, takes place in blends with low solid mass fraction, a high kinetic barrier is established. As a consequence, the growth of nanocrystals is restricted leading to the formation of predominantly small nanoparticles. On the other hand at high solid mass fraction (and therefore high viscosities) the molecules have already a low mobility in the fat matrix and hence a restricted growth of the nanocrystals. When rapidly cooling from the melt, the generated kinetic barrier must be added to the high existing barrier; as a consequence the effect of high cooling rate, expressed as smaller nano-platelets is not pronounced.

Cryo-TEM micrographs of the blends of FHCO and canola oil. Images correspond to 50% of FHCO (A–B) and 100% of FHCO (C–D), crystallized at 0.7°C/min (A and C), and 10°C/min. (B–D).
Rheological properties
To investigate the influence of composition and nanostructure of the crystalline network on the mechanical properties of the network the shear storage modulus of FHCO and also binary mixtures of FHCO and canola oil crystallized at cooling rates of 10 and 0.7°C/min were determined.
Plots of G′ versus SFC of all the samples crystallized under both fast and slow cooling rates are presented in Fig. 5. This figure reports a clear relationship between SFC and G′ of the blends when high correlation coefficients (0.84< r2 <0.98) between the storage modulus (G′) and SFCs under both crystallization conditions.

Shear storage modulus as a function of SFC (obtained from different dilutions of FHCO and canola oil) at 0.7°C/min and 10°C/min cooling rates.
Increasing the stearic acid ratio, a saturated fatty acid with high melting point, was positively correlated with increasing G′ in the range studied. However, an exception to this behavior was observed for 100% FHCO. Even though pure FHCO has the highest amount of solids (98%), it has the lowest shear storage modulus. This is most likely due to the presence of the alpha polymorphic form in this sample.
Using the slope of ln-ln plots of the elastic modulus versus volume fractions (Φ) the fractal dimension (D) for the distribution of fat crystal particles within the network was determined. A higher value of fractal dimension (D = 2.15) was obtained when the blends are crystallized at lower cooling rate compared to D = 2.0 for crystallization at 10°C/min.
Moreover, as shown in this figure, a higher elastic modulus was observed for the samples crystallized at 10°C/min compared to the samples crystallized under slow cooling conditions. These results demonstrate that samples with similar SFCs and polymorphism can have very different hardnesses, which is attributable to differences in structure at the nano and mesoscales. In order to explore this phenomenon, crystal morphology at both meso and nanoscales were characterized under the different cooling rate conditions (shown in Figs. 3 and 4). Even though in most fat blends, a spherulitic crystal morphology was observed at every cooling rate, the formation of a less dense matrix in the slow cooled samples was evident. As expected, the crystal network in fast-cooled samples displayed a larger number of smaller spherulites compared to those crystallized at slow cooling rates (Fig. 3). This difference in spherulite size is a consequence of the high nucleation density at high cooling rates, promoted in turn, by the pronounced dynamic supercooling generated during the crystallization process. Figure 6A reports the meso-structural elements' equivalent diameters obtained from image analysis of the PLM micrographs. Blends crystallized from the melt under rapid cooling conditions showed mesocrystals with smaller equivalent diameters (p < 0.05) than those formed under slow cooling conditions. As mentioned before, slow cooling leads to formation of fewer nuclei that are able to grow and aggregate into larger crystalline structures. The results show that a faster cooling rate induced a decrease in crystal dimensions between 35 and 60%. It is important to note that in slowly cooled blends, crystal equivalent diameters decreased with increasing canola oil proportions (p < 0.05). In line with our observations, Rousseau et al. 28 reported crystal diameter increases while increasing the liquid oil proportion in fat blends of palm oil/soybean oil and lard/canola oil. Ribeiro et al. 7 have also observed similar trends for fully hydrogenated soybean oil diluted with soybean oil. On the other hand, this effect of supersaturation was not observed at high cooling rates where no significant differences in crystal size were observed between the different hardstock proportions.

Equivalent diameters of the meso- (A) and nano- (B) structural elements as a function of FHCO proportion (%FHCO) obtained from blends crystallized at slow (0.7°C/min), and fast cooling rates (10°C/min). Different superscript letters represents significantly different calculated diameters (p < 0.05).
Figure 6B shows the changes in the nano-particle equivalent diameters observed in blends with 50, 80, and 100% of FHCO crystallized at different cooling rates. Similar to the meso-scale, nano crystals size decreased when cooling rate was increased from 0.7 to 10°C/min. For instance, the equivalent diameter of nano-platelets in 50% FHCO were 116 nm and 240 nm for fast and slow cooling rates, respectively. It is worth noting that size changes were less evident in blends with 100% of hardstock, however the nano-plateles were still significantly larger (p < 0.05) in the slowly cooled samples, compared to those found in rapidly cooled mixtures. Additionally, a significant decrease (p < 0.05) in nanoplatelet sizes, induced by the increase in supersaturation, was observed at both crystallization conditions.
The data in Fig. 6 shows an opposite behavior between meso and nano-particle dimensions as a function of FHCO dilution (changes in supersaturation). On the other hand, both structural levels exhibited that particle size decrease when the cooling rate increases. Furthemore, it seems that the nanoscale is less sensitive to the cooling rate than meso-structure since for instance at 10°C/min there is no effect of supersaturation on meso-crystal size, which may be the result of the high impact on meso-structure of the cooling rate increase. Meanwhile at the nano-scale is likely that the effect of increased cooling rate is not so important which results in also evident effects of supersaturation alteration in the systems.
All these results provide strong evidence of the effects of cooling rate on the crystalline structure of fats, namely crystal size at both micro and nano scales, regardless of polymorphic form.
 ((2))
((2)) ((3))
((3))Fractal dimension, D, was determined by the slope of a ln–ln plot of elastic modulus (G′) versus volume fraction Φ according to Narine and Marangoni 30.
Using the fractal dimensions of the samples at both fast and slow cooling (D10 = 2.0 and D0.7 = 2.15), the elastic modulus (G′) and the volume fraction of the solid in the blends, the ratio of λ as well as the ratio of the samples elastic modulus at the applied cooling rates were calculated and reported in Table 3. Similar to the G′ ratio, a higher λ parameter is obtained in the samples crystallized under fast cooling rates relative to the sample crystallized under slow cooling conditions. As shown in Table 1, the applied slow and fast cooling rates did not affect the samples' SFC significantly and the ratio of SFCs were equal to 1 (see Table 3). Interestingly, the ratio of storage moduli were comparable to the ratio of Φ1/3–D under the same conditions. This result confirms that the differences in the rheological properties of the samples are not only dependent to the solid mass fraction, but also the spatial distribution of crystals and their interaction within the network. A lower fractal dimension for the sample crystallized at 10°C/min versus the sample crystallized at 0.7°C/min would translate into a higher elastic modulus. To further investigate the relationship between the networks' elasticity and the particles characteristics, we also calculated the ratio of equivalent diameters of the samples at both the nano and meso scales (shown in Table 3). It is important to note that the different cooling rates affected the nano-structure and microstructure of the blends to a great extent. The ratio of crystal sizes at the different cooling rates was higher than the ratios of Φ1/3–D or λ. This finding suggests that an important factor within λ must have been significantly affected by cooling rate in order to mitigate crystal size effects. This analysis points to possibly intermolecular interactions, via the Hamaker constant, or inter-crystalline separation distance. Put in another way, the crystal melt interfacial tension must be lower for samples crystallized at high cooling rates versus low cooling rates. This actually makes sense since at high cooling rates, molecular mixing in the solid state is enhanced, which would decrease the differences in chemical nature between the solid and the liquid.
| FHCO (%) | 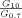 |
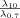 |
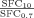 |
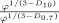 |
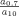 |
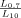 |
|---|---|---|---|---|---|---|
| 50 | 1.20 | 1.08 | 1 | 1.18 | 2.04 | 2.22 |
| 60 | 1.27 | 1.17 | 1 | 1.15 | 1.63 | 1.41 |
| 70 | 1.10 | 1.1 | 0.99 | 1.08 | 1.44 | 2.17 |
| 80 | 1.07 | 1.04 | 1 | 1.06 | 1.33 | 1.72 |
| 90 | 1.10 | 1.1 | 0.99 | 1.02 | 1.26 | 2.27 |
| 100 | 0.59 | ND | 1 | ND | 1.09 | 2.44 |
Attempts to affect the polymorphic forms of FHCO
As explained above none of the applied crystallization cooling rates modified the structure of 100% FHCO. Therefore, to better understand the effect of cooling rate on the kinetics and physical properties of pure FHCO, this sample was also crystallized at 0.1°C/min. Pulse-NMR analysis showed similar amount of solid (98%) in pure FHCO crystallized under different cooling rates. XRD profiles and DSC patterns of pure FHCO crystallized under the three different cooling rates (0.1, 0.7, and 10°C/min) are presented in Fig. 7. Figure 7A displays three different melting peaks for 100% FHCO crystallized at higher cooling rates including 10 and 0.7°C/min. These melting points are correlated with a transition of the less stable polymorph form into a more stable phase during the melting process. The first peak belongs to α-form and the second and third peaks to β′ and β forms, respectively. However, when FHCO is crystallized at 0.1°C/min, only two sharp melting peaks corresponding to β′ and β polymorphs are observed. Since the β′→ β transition form can occur during the heating process, a more accurate estimate of the phase behavior was obtained using XRD, Fig. 7B. Based on the X-ray analysis, the α-form can be considered as the main polymorphic phase of FHCO crystallized at high cooling rates. Crystallization under slower cooling rates promoted the formation of the β-polymorph.

(A) Melting profile of 100% FHCO crystallized at different cooling rates., (B) XRD pattern of 100% FHCO crystallized by fast cooling rate (10°C/min), and 0.1°C/min at 20°C.
As shown in Fig. 8, the shear modulus increased as the cooling rate decreased and based on statistical analysis the shear modulus value of these three different cooling rates are significantly different (p < 0.05). These results are evidence of the weaker structure of the α-polymorphic form of FHCO relative to the β-form. This study introduces another alternative method to produce the β-crystal forms with more functionality. However, a great challenge remains to stabilize the alpha polymorphic form.

Shear storage modulus (G′) of 100% FHCO crystallized at 20°C with different cooling rate. Superscript letters represents statistically significant differences between the shear storage modulus values.
Conclusion
Here we have shown the influence of the processing conditions and blending with liquid canola oil on the nano/microstructure and rheological properties of FHCO. We observed different macroscopic properties, structural characteristics and polymorphic forms in FHCO crystallized at high and low cooling rates and within a diverse range of oil dilutions. Interestingly, we found that the structure of a fat is affected differently at different length scales, depending on the nature of the external field (temperature, concentration, shear). This also indicates the need to monitor both the nanoscale and mesoscale in fats in order to explain and engineer their functionality.
The amount of solid fat was usually sufficient to predict the hardness of the fat blends. However, oscillatory rheology demonstrated that the α-polymorphic form was mechanically weaker than the beta polymorphic form. This effect could be due to the presence of larger microstructural elements present with fewer bonds to strengthen the fat crystal network. It may also suggest the agglomeration of the small nano-particles of α-polymorph, resulting in formation of larger clusters which affect the mechanical properties of the system.
Moreover, here it is shown that solution behavior was highly affected by varying the SFC, which was determined by the Hildebrand model. Deviations from the ideal behavior occurred at higher dilutions probably due to the formation of solid solutions or imperfect crystals form, while they have a greater solubility than perfect crystals.
Our results show that blending with liquid oil is a valuable tool which can be used for the modification of the rheological properties of FHCO. Blending leads to changes in the hardness and solid-like character of FHCO by inducing changes in the polymorphic form and therefore in the structure of the fat crystal network without affecting the amount of solids present.
The authors have declared no conflict of interest.

