

Allylic oxidation of methyl 10-undecenoate and nucleophilic additions to methyl 9-oxo-10-undecenoate
Abstract
Methyl 10-undecenoate (2b) has been oxidized with tert-butyl hydroperoxide/selenium dioxide and potassium dichromate to 53% of methyl 9-oxo-10-undecenoate (4). Based on the conversion of 2b, the yield of 4 is 81%. Residual selenium in ester 5 was determined to be less than 1 ppm. The electrophilic double bond in enone 4 reacts in good to excellent yield with a variety of nucleophiles. With dimethyl malonate, acetylacetone, and methyl acetoacetate Michael–adducts at C-11 of enone 4 were obtained in 88–99% yield. Corresponding additions were achieved with nitroethane, 1- and 2-nitropropane in 78–89% yield. In a Nef–reaction some nitroalkyl adducts were converted to methyl esters of 9,12-dioxofatty acids in 99% yield. In methanol/sodium methoxide the ester methyl 11-methoxy-9-oxoundecanoate was obtained in 86% yield from enone 4. In a three step reaction with ammonium chloride/sodium cyanide, hydrochloric acid/acetic acid, and methanol/2,2-dimethoxypropane the ester 4 yielded 63% of dimethyl 4-oxo-dodecanedioate. With the cyanide ion the electrophilic carbon atom in aldehydes can be converted into a nucleophile (Stetter–reaction). Catalysed by sodium cyanide in DMF aromatic and heteroaromatic aldehydes RCHO with R = phenyl, 2-thienyl, 2-furyl and 3-pyridyl were added to enone 4 to afford methyl 12-aryl- and methyl 12-heteroaryl-9,12-dioxododecanoates in 54–73% yield.
Practical applications: The prepared oleochemicals are useful as precursors for pharmaceuticals and as monomers for polyesters and polyamides. They also may be applied to synthesize fatty acid conjugates by providing suitable connectors for biological active compounds and to attach groups with suitable physical properties for materials.
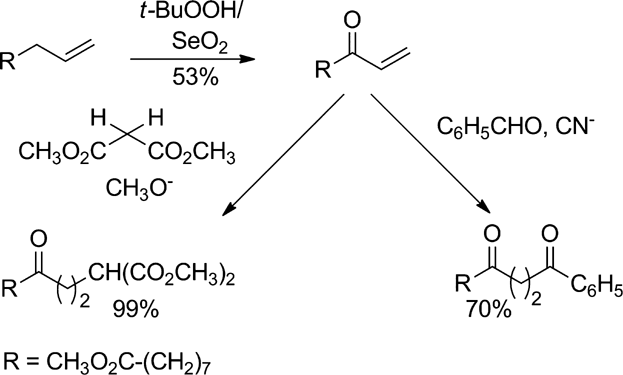
For enone 4 prepared from methyl 10-undecenoate, thirteen Michael additions with 1,3-dicarbonyl and nitro alkyl compounds, hydrocyanic acid and acyl anions are reported.
1 Introduction
Fatty acids are abundant and renewable raw materials. They are often essential parts in surfactants 1, 2, cosmetic and food additives 3, lubricants 4, corrosion inhibitors 5 and antioxidants 3, 6. Furthermore, they gain increasing interest as renewable monomers for polymerisation 7, 8. They can be connected in fatty acid conjugates with bioactive compounds; thereby existing antioxidant, antimicrobial, antiviral or anticancer activities are increased 9, 10. Furthermore, these conjugates assist in drug targeting of bioactive compounds by improving their interaction with the cell membranes and their transmembrane delivery 11-13.
A versatile intermediate to synthesize compounds for applications named in ref. 1-13 could be 10-undecenoic acid (2a). The unsaturated fatty acid 2a is accessible from castor oil, a vegetable oil, being produced in India, China and Brazil in together more than one million tonnes in 2007; the major component (90%) is ricinoleic acid (1, Figure 1). Technical applications of castor oil are dehydration to octadecadienoic acids, hydrogenation, sulfonation, pyrolysis to 10-undecenoic acid (2a) and heptaldehyde, and thermal alkali fission to 2-octanol and sebacic acid 14.

The polarity of the nucleophilic double bond in methyl ricinoleate can be transformed into an electrophile, which allows the addition of nucleophilic reagents. This has been demonstrated by the oxidation of the hydroxyl group at C-12 of the ester of 1b to a ketone and the base catalysed conjugation of the double bond to methyl (E)-12-oxo-10-octadecenoate (3). Enone 3 allows the addition of synthetically useful nucleophiles at the internal C-10 atom 15.
Methyl 10-undecenoate (2b) has been used for radical ene-additions at the terminal double bond leading to oleochemicals of synthetic interest 16. If one succeeds to introduce selectively a carbonyl group at the C-9 atom of ester 2b one has generated an enone with an electrophilic double bond that allows the addition of nucleophilic reagents at the terminal carbon atom C-11. This should widen the range of synthetically useful structures in oleochemistry and give insight into the positional reactivity of internal and terminal enones of fatty acids. At first we will report on the possibility to convert methyl 10-undecenoate (2b) into methyl 9-oxo-10-undecenoate (4). Thereafter, we describe the reaction of 4 with a number of nucleophiles.
2 Materials and methods
2.1 Analytical equipment
FT-IR-Spectra were recorded with a 5 DXC-FT-IR-spectrometer (Nicolet). 1H NMR- and 13C NMR-spectra were measured with a WM 300 spectrometer (Bruker) at 300 and 75.5 MHz. Mass spectra (EI, 70 eV) were obtained with a MAT 312 instrument (Finnigan - MAT). For GC-MS coupling the gaschromatographs 1400, 3400 (Varian) and GC-8A (Shimadzu) were combined with mass spectrometers MAT CH-7a and MAT 8230 (Varian, Finnigan) and Varian Saturn II; the last one works with an ion trap that amplifies ions with longer life, these spectra are marked with an asterisk.
TLC was performed with silicagel 60 F254 (Merck). For flash chromatography silicagel 60 [70–230 mesh] (Merck) was used. Elemental analyses were performed by the analytic laboratory of the Institute of Organic Chemistry of the University of Münster and the microanalytical laboratory M. Beller (Göttingen). Melting points are uncorrected. Yields refer to the substance used as minor component. All reactions, where oxygen and water had to be excluded, were done under argon in baked-out glass vessels sealed with a septum. Reagents and solvents were added with microliter syringes (TLL 1000, Hamilton). For the atom absorption spectroscopy the instrument 4100 ZL (Perkin Elmer) was used.
2.2 Chemicals
The employed chemicals (for their purity see Supporting information: 2.2 Chemicals) were purchased from Aldrich, Merck, and Sigma and were used if not stated otherwise without further purification. All solvents were purified by distillation and if necessary residual water was removed. The composition of solvents and eluents are given in volume ratios of the components.
2.3 Experimental procedures
2.3.1 Preparation of methyl 9-oxo-10-undecenoate (4)
2.3.1.1 Methyl 10-undecenoate (2b)
Undecenoic acid (46.1g, 0.25 mol) was dissolved in dry methanol (10 mL), 2,2-dimethoxypropane (61 mL, 0.5 mol) and conc. hydrochloric acid (0.5 mL) and stirred for 24 h at RT. Thereafter, the solvent was rotaevaporated and the residue distilled at the vacuum of the water pump to afford the ester 2b (47.4 g, 0.24 mol) as colorless oil. n20D = 1.4391(n20D = 1.4393 17.
2.3.1.2 Allyloxidation of methyl 10-undecenoate (2b) 18, 19
Tert-butyl hydroperoxide (2.04 mL, 20 mmol, 80% in di-tert-butylperoxide) are dissolved in dichloromethane (20 mL), and selenium dioxide (0.55 g, 5 mmol) is added. After stirring for 0.5 h at RT ester 2b (1.98 g, 10 mmol) in dichloromethane (5mL) is added dropwise. After stirring for 24 h the solution is concentrated by rotaevaporation, then water (50 mL) is added and the mixture is extracted with petroleum ether (4 × 25mL). The organic phase is dried (MgSO4) and then rotaevaporated and subjected to flash chromatography (petroleum ether/diethyl ether = 5:3) to afford 50–55% of hydroxy ester 5 (1.07–1.18 g, 5.0–5.5 mmol) and 7–12% of enone 4 (0.15–0.25 g, 0.7–1.2 mmol) as colorless oils.
A sample of ester 5 (0.5 g) was refluxed for 8 h in conc. nitric acid (30 mL) until a homogeneous solution was obtained. The solution was filled up to 100 mL and investigated by atom absorption spectroscopy (AAS). Within the detection limit (0.1 ppb) no selenium was found; calculated for the sample the content is maximal 1 ppm.
Methyl 9-hydroxy-10-undecenoate (5): Rf-value: 0.24 (petroleum ether/diethyl ether = 5:2).- 1H NMR (CDCl3): δ (ppm) = 1.24 (m, 8H, CH2), 1.44 (m, 2H, CH2CH(OH)), 1.53 (m, 2H, CH2CH2CO), 2.23 (t, J = 7.5 Hz, 2H, CH2CO), 3.59 (s, 3H, OCH3), 4.02 (m, 1H, CH(OH)), 5.02 (ddd as dt, J = 1.3 Hz, Jcis = 10.6 Hz, 1H, CHCHaHb), 5.14 (ddd as dt, J = 1.5 Hz, Jcis = 17.2 Hz, 1H, CHCHaHb), 5.79 (ddd, J = 6.3 Hz, Jcis = 10.6 Hz, Jtrans = 17.2 Hz, 1H, CHCH).- The spectroscopic data agree well with those in ref. 19, 20.
Methyl 9-oxo-10-undecenoate (4): Rf-value: 0.35 (petroleum ether/diethyl ether = 5: 2).- 1H NMR (CDCl3): δ (ppm) = 1.24 (m, 8H, CH2), 1.54 (m, 4H, CH2CH2CO), 2.22 (t, J = 7.5 Hz, 2H, CH2CO2), 2.49 (t, J = 7.4 Hz, 2H, CH2CO), 3.59 (s, 3H, OCH3), 5.73 (dd, Jgem = 1.5 Hz, Jcis = 10.2 Hz, 1H, CHCHaHb), 6.13 (dd, Jgem = 1.5 Hz, Jtrans = 17.7 Hz, 1H, CHCHaHb), 6.28 (dd, J = 6.3 Hz, Jcis = 10.2 Hz, Jtrans = 17.7 Hz, 1H, CHCH2).-
The spectroscopic data agree well with these in ref. 21.
2.3.1.3 Oxidation of methyl 9-hydroxy-10-undecenoate (5)
To hydroxyester 5 (2.12 g, 10 mmol) dissolved in diethyl ether (60 mL) under ice-cooling and strong stirring within 10 min an acidic dichromate solution (16 mL), consisting of potassium dichromate (6 g, 20.5 mmol) dissolved in water (38 mL) and conc. sulphuric acid (9.5 mL) is added. Thereafter, stirring is continued for 40 min at 0 °C until 5 is totally converted (TLC control) and then 10% aqueous hydrochloric acid (2 mL) is added. Excess chromium reagent is reduced with a saturated sodium bisulphite solution. Thereafter, the organic phase is removed and the aqueous phase is extracted with diethyl ether (3 × 25 mL). The combined organic phases are washed with a 10% sodium ethylenediaminetetraacetate solution being adjusted with ammonia to pH 7 (2 × 10 mL). After drying (sodium sulphate) the solution of the crude product is rotaevaporated and the residue purified by flash chromatography (petroleum ether/diethyl ether = 5:2). Thereby, 84% of enone 4 (1.78 g, 8.40 mmol) is obtained as colorless oil.
2.3.2 Reactions of methyl 9-oxo-10-undecenoate (4)
2.3.2.1 Reaction with C-H acidic compounds
2.3.2.1.1 Addition of β-dicarbonyl compounds
2.3.2.1.1.1 General procedure for the addition of ß-dicarbonyl compounds 22
In an annealed round bottom flask sodium was reacted with methanol and then the five-fold molar amount of the 1,3-dicarbonyl compound was added. Thereafter, in an amount being equimolar to the amount of sodium enone 4 in dry methanol (5 mL) is added dropwise and then the solution is stirred for several hours at RT. Then water (20 mL) is added and extracted with diethyl ether (3 × 20 mL). The combined organic phases are dried (MgSO4) and the solvent rotaevaporated. Finally, the product is filtered through a short column and dried at an oil-diffusion pump.
2.3.2.1.1.2 Dimethyl 12-methoxycarbonyl-9-oxotridecanedioate (7)
According to the general procedure in 2.3.2.1.1.1, enone 4 (0.20 g, 0.94 mmol) and dimethyl malonate (6, 0.62 g, 4.7 mmol) are reacted at RT with continued stirring for 5 h to afford 7 (0.32 g, 0.93 mmol, 99%) as a colorless oil that solidifies.- Rf-value: 0.31 (petroleum ether/diethyl ether = 1:1).- M.p.: 44–45 °C.- 1H-NMR (CDCl3): δ (ppm) = 1.17 (m, 6H, CH2), 1.45 (m, 4H, CH2CH2CO), 2.02 (dt, J =7.2 Hz, 2H, CH2CH(CO2CH3)2), 2.16, 2.26, 2.38 (3t, J = 7.4 Hz, J = 7.4 Hz, J = 7.4 Hz, 6H, CH2CO), 3.31 (t, J = 7.4 Hz, 1H, CH(CO2CH3)2), 3.53 (s, 3H, OCH3), 3.60 (s, 6H, CH(CO2CH3)2).- C17H28O7 (344.40): calc. C 59.29, H 8.19; found: C 59.10, H 8.30.
2.3.2.1.1.3 Methyl 9,13-dioxo-12-methylcarbonyltetradecanoate (9)
According to the general procedure in 2.3.2.1.1.1, enone 4 (0.24 g, 1.16 mmol) and acetylacetone (8, 0.80 g, 8.0 mmol) are reacted at RT with stirring for ten hours. Work-up afforded a colorless oil that led by cooling to 98% of 9 (0.49 g, 1.57 mmol) as white solid.- Rf-value: 0.33 (petroleum ether/diethyl ether = 1:2).- M.p.: 29–30 °C.- 1H NMR (CDCl3): δ (ppm) = 1.20 (m, 6H, CH2), 1.35-1.6 (m, 4H, CH2CH2CO), 1.97 (dt, J = 6.9 Hz, 2H, CH2CH(COCH3)2), 2.04 (s, 0.34 × 6H = 2H, H3CC(OH) = CCOCH3, enol tautomer), 2.09 (s, 0.66 × 6H = 4H, H3CCOCHCOCH3, keto tautomer), 2.19, 2.27, 2.31 (3t, J = 7.4 Hz, J = 7.4 Hz, J = 6.8 Hz, 6H, CH2CO), 3.56 (s, 3H, OCH3), 3.58 (t, J = 7.7 Hz, 0.66 H, CH(COCH3)2, keto tautomer), 15.94 (s, 0.34 H, C = C(CH)OH, enol tautomer).- C17H28O5 (312.41): calc. C 65.36, H 9.03; found: C 65.19, H 9.26.
2.3.2.1.1.4 Dimethyl 12-methylcarbonyl-9-oxotridecanedioate (11)
According to the general procedure in 2.3.2.1.1.1, enone 4 (0.24 g,1.16 mmol) was added at −30 °C to a solution of methyl acetoacetate (10, 0.66 g, 5.70 mmol) in methanol. Then the solution was cooled to −30 °C and the reaction was continued at −30 °C with stirring for 12 h. Work-up with flash chromatography (petroleum ether/diethylether = 1:1) afforded a colorless oil that solidified after prolonged standing to 11 as a white solid (0.34 g, 1.02 mmol, 88%).- Rf-value:0.24 (petroleum ether/diethylether = 1:1).- M.p.: 19–21 °C.- 1H NMR (CDCl3): δ (ppm) = 1.16 (m, 6H, CH2), 1.43 (m, 4H, CH2CH2CO), 1.93 (m, 2H, diastereotopic 11-CH2), 2.09 (s, 3H, H3CCO), 2.15, 2.24, 2.32 (3t, J = 7.5 Hz, J = 7.5 Hz, J = 7.2 Hz, 6H, CH2CO), 3.38 (t, J = 7.2 Hz, 1H, H3CCOCHCO2CH3), 3.51, 3.59 (2s, 6H, OCH3).- C17H28O6 (328.40): calc: C 62.17, H 8.59; found: C 61.94, H 8.87.
2.3.2.1.1.5 Methyl 8-(3′-cyclohex-2′-enonyl)octanoate (12)
In an annealed round bottom flask diester 11 (0.26 g, 0.79 mmol) was dissolved in dry tetrahydrofuran (70 mL) that contained sodium metal (10 mg, 0.43 mmol); the solution was then refluxed for 7 h. Thereafter, water (50 mL) was added. The organic phase was separated and the aqueous phase was extracted with diethyl ether (3 × 15 mL). The combined organic phases were dried (MgSO4) and the solvent rotaevaporated. After a column filtration (petroleum ether/diethyl ether = 1: 2) product 12 (0.16 g, 0.63 mmol, 80%) was obtained as colorless oil.- Rf-value: 0.18 (petroleum ether/diethyl ether = 1:1).- 1H NMR (CDCl3): δ (ppm) = 1.20 (m, 6H, CH2), 1.38 (m, 2H, CH2), 1.50 (m, 2H, CH2CH2CO), 1.86 (m, 2H, 5′-CH2), 2.09 (t, J = 7.3 Hz, 2H, 8-CH2), 2.14–2.28 (3t, J = 6.8 Hz, J = 7.5 Hz, J = 6.6 Hz, 6H, CH2CO2, 4′-/6′-CH2), 3.54 (s, 3H, OCH3), 5.74 (m, 1H, CCH).- C15H24O3 (252.35): calc.C 71.39, H 9.59; found: C 71.42, H 9.51.
2.3.2.1.2 Addition of nitroalkanes 21
2.3.2.1.2.1 General procedure for the addition of nitroalkanes
An annealed round bottom flask was charged under argon with sodium (41 mg, 1.8 mmol) in methanol (10 mL). Then, the nitroalkane (5 mmol) was added. Thereafter, within 10 min enone 4 (0.32 g, 1.5 mmol) in dry methanol (10 mL) was added dropwise with stirring, which was continued for 3 h. For work up water (30 mL) was added and the mixture was extracted with diethyl ether (3 × 30 mL). After drying the organic phase (MgSO4) the solvent was rotaevaporated and the crude product was purified by flash chromatography (petroleum ether/diethyl ether = 5:3). Compounds 14–16 were obtained as colorless oils.
2.3.2.1.2.2 Methyl 12-nitro-9-oxotridecanoate (14)
Enone 4 (0.32 g, 1.5 mmol) and nitroethane (0.38 g, 5 mmol) afford 14 (0.33 g, 1.17 mmol, 77%).- Rf-value: 0.26 (petroleum ether/diethyl ether = 5:3).- 1H NMR (CDCl3): δ (ppm) = 1.19 (m, 6H, CH2), 1.3–1.6 (m, 4H, CH2CH2CO), 1.42 (d, J = 6.4 Hz, 3H, CHNO2CH3), 1.99 (m, 2H, CH2CHNO2), 2.18, 2.29 (2t, J = 7.5 Hz, J = 7.4 Hz, 4H, CH2CO), 2.37 (t, J = 7.0 Hz, 2H, 10-CH2), 3.54 (s, 3H, OCH3), 4.49 (m, 1H, CHNO2).- C14H25NO5 (287.33): calc. C 58.51, H 8.77, N 4.88; found: C 58.39, H 8.63, N 5.01.
2.3.2.1.2.3 Methyl 12-nitro-9-oxotetradecanoate (15)
Enone 4 (0.32 g, 1.5 mmol) and 1-nitropropane (0.45 g, 5 mmol) afford 15 (0.40 g, 1.33 mmol, 89%).- Rf -value: 0.36 (petroleum ether/diethyl ether = 5:3).- 1H NMR (CDCl3): δ (ppm) = 0.88 (t, J = 7.3 Hz, 3H, CH2CH3), 1.22 (m, 6H, CH2), 1.50 (m, 4H, CH2CH2CO), 1.6–2.1 (m, 4H, CHaHbCHNO2CHcHd), 2.21, 2.31(2t, J = 7.4 Hz, J = 7.5 Hz, 4H, CH2CO), 2.38 (t, J = 7.0 Hz, 2H, 10-CH2), 3.57 (s, 3H, OCH3), 4.35 (m, 1H, CHNO2).- C15H27NO5 (301.38): calc. C 59.78, H 9.03, N 4.65; found: C 59.66, H 9.17, N 4.91.
2.3.2.1.2.4 Methyl 12-methyl-12-nitro-9-oxotridecanoate (16)
Enone 4 (0.32 g, 1.5 mmol) and 2-nitropropane (0.45 g, 5 mmol) afford 16 (0.40 g, 1.33 mmol, 89%). Rf-value: 0.27 (petroleum ether/diethyl ether = 5:3).- 1H NMR (CDCl3): δ (ppm) = 1.11 (m, 6H, CH2), 1.3–1.5 (m, 4H, CH2CH2CO), therein 1.39 (s, 6H, CH3), 1.99 (t, J = 7.4 Hz, 2H, 11-CH2), 2.10 (t, J = 7.3 Hz, 2H, CH2CO), 2.23 (m, 4H, CH2CO), 3.45 (s, 3H, OCH3).- C15H27NO5 (301.38): calc. C 59.78, H 9.03, N 4.65; found: C 59.57, H 9.15, N 4.68.
2.3.2.1.3 Nef-reaction of the nitro compounds 21
2.3.2.1.3.1 General procedure for the Nef-reaction
A solution of the nitro compound in methanol (1 mL) is added dropwise at RT to the equimolar amount of sodium in methanol (10 mL). The suspension is added dropwise at −10 °C within 30 min to conc. sulfuric acid (0.23 mL) in methanol (0.6 mL) and is then stirred for 30 min. To the reaction mixture water (30 mL) is added and then the mixture is extracted with diethyl ether (3 × 15 mL). After drying (MgSO4) the solvent is rotaevaporated. The product is obtained after flash chromatography (petroleum ether/diethyl ether = 1:1) as white solid.
2.3.2.1.3.2 Methyl 9,12-dioxotridecanoate (18)
14 (0.25 g, 0.87 mmol) affords 18 (0.22 g, 0.86 mmol, 99%).-Rf-value: 0.25 (petroleum ether/diethyl ether = 1:1.- M.p.: 27 − 28°C.- 1H NMR (CDCl3): δ (ppm) = 1.21 (m, 6H, CH2), 1.49 (m, 4H, CH2CH2CO), 2.08 (s, 3H, O = CCH3), 2.19, 2.35 (2t, J = 7.3 Hz, J = 7.3 Hz, 4H, CH2CO), 2.59 (m, 4H, O = CCH2CH2CO), 3.56 (s, 3H, OCH3).- C14H24O4 (256.35): calc. C 65.60, H 9.44; found: C 65.64, H 9.30.
2.3.2.1.3.3 Methyl 9,12-dioxotetradecanoate (19)
15 (0.3 g, 1 mmol) affords 19 (0.27 g, 0.99 mmol, 99%).- Rf–value: 0.41 (petroleum ether/diethyl ether = 1:1).- M.p.: 31 − 32°C.- 1H NMR (CDCl3): δ (ppm) = 0.90 (t, J = 7.4 Hz, 3H, CH2CH3), 1.16 (m, 6H, CH2), 1.44 (m, 4H, CH2CH2CO), 2.14 (t, J = 7.3 Hz, 2H, CH2CO2), 2.32 (m, 4H, 8-CH2, 13-CH2), 2.52 (s, 4H, O = CCH2CH2CO), 3.50 (s, 3H, OCH3).- C15H26O4 (270.37): calc. C 66.63, H 9.69; found: C 66.26, H 9.63.
2.3.2.1.4 Addition of methanol [see supporting information 2.3.2.1.4]
2.3.2.2 Preparation of dimethyl 4-oxododecanedioate (23)
2.3.2.2.1 Methyl 11-cyano-9-oxoundecanoate (21)
A solution of ammonium chloride (0.13 g, 2.38 mmol) and potassium cyanide (0.19 g, 3 mmol) in water (1 mL) was dropped to a solution of enone 4 (0.42 g, 2 mmol) in DMF (2.5 mL), the mixture was heated for 50 min to 90 °C. Then after cooling to RT, water (50 mL) is added. After extraction with diethyl ether (3 × 20 mL) the combined organic phases are washed with water and a sat. sodium chloride solution and dried (MgSO4). Flash chromatography (petroleum ether/diethylether = 1:2) affords 21 (0.39 g, 1.63 mmol, 82%) as white solid.- Rf-value: 0.33 (petroleum ether/diethyl ether = 1:2).- M.p.: 44–45 °C.- 1H NMR (CDCl3): δ (ppm) = 1.23 (m, 6H, CH2), 1.51 (m, 4H, CH2CH2CO), 2.21 (t, J = 7.5 Hz, 2H, CH2CO), 2.39 (t, J = 7.4 Hz, 2H, 8-CH2), 2.49 (t, J = 7.0 Hz, 2H, 10-CH2), 2.72 (t, J = 7.0 Hz, 2H, CH2CN), 3.57 (s, 3H, OCH3).- C13H21NO3 (239.31): calc. C 65.25, H 8.84, N 5.85; found: C 65.23, H 9.10, N 5.95.
2.3.2.2.2 4-Oxododecanedioic acid (22)
21 (0.37 g, 1.55 mmol) is converted to the white solid 22 (0.35 g, 1.43 mmol, 92%) using a procedure given in ref. 23.- Rf–value: 0.15 (petroleum ether/diethyl ether/acetic acid = 1:2:0.01).- M.p. 109 − 110°.- 1H NMR (MeOD): δ (ppm) = 1.33 (m, 6H, CH2), 1.58 (m, 4H, CH2CH2CO), 2.28, 2.48 (2t, J = 7.3 Hz, J = 7.2 Hz, 4H, 5-/11-CH2), 2.52, 2.72 (2t, J = 6.4 Hz, J = 6.4 Hz, 4H, 2-/3-CH2), 4.99 (br. s, 2H, OH).- C12H20O5 (244.29): calc. C 59.00, H 8.25; found C 59.06, H 8.20.
2.3.2.2.3 Dimethyl 4-oxododecanedioate (23)
22 (0.6 g (1.55 mmol) was dissolved in dry methanol (5 mL). Then 2,2-dimethoxypropane (5 mL) and one drop conc. hydrochloric acid was added and the mixture was stirred at RT for 24 h. Thereafter, the solvent was rotaevaporated and the residue was purified by flash chromatography to afford 83% of 23 (0.35 g, 1.29 mmol) as colorless oil.- 1H NMR (CDCl3): δ (ppm) = 1.20 (m, 6H, CH2), 1.48 (m, 4H, CH2CH2CO), 2.18 (t, J = 7.5 Hz, 2H, CH2CO2), 2.33, 2.46, 2.61 (3t, J = 7.3 Hz, 6.5 Hz, 6.5 Hz, 6H, CH2COCH2CH2CO2), 3.54, 3.55 (2s, 6H, OCH3).- The spectroscopic data agree well with those in the literature 23-25.
2.3.2.3 Reaction with aromatic aldehydes
2.3.2.3.1 General procedure for the addition of aromatic aldehydes 26
A solution of freshly distilled aldehyde (2.67 mmol) in dry DMF (2 mL) is added at 35 °C under stirring within 10 min to sodium cyanide (13 mg, 0.27 mmol) in DMF (2 mL), stirring is continued for 30 min. Thereafter, within 30 min at 35 °C enone 4 (0.42 g, 2 mmol) in DMF (4 mL) is added. After stirring for 1 h at 35 °C water (50 mL) is added and then the mixture is extracted with diethyl ether (3 × 15 mL). The combined organic phases are washed with diluted sulphuric acid; then the organic phase is washed with a diluted sodium hydrogen carbonate solution until the aqueous phase is neutral. After drying the organic phase (MgSO4) the solvent is rotaevaporated and the crude product purified by flash chromatography. All adducts are obtained as colorless solids, which, however, change colour on standing. The hydrogen cyanide adduct 21 is obtained in all reactions as side product.
2.3.2.3.2 Methyl 9,12-dioxo-12-phenyldodecanoate (24)
Enone 4 (0.42 g, 2 mmol) and benzaldehyde (0.28 g, 2.67 mmol) afford 70% 24 (0.45 g, 1.41 mmol).- Rf–value: 0.29 (petroleum ether/diethyl ether = 5:3).- M.p.: 40–41 °C.- 1H NMR (CDCl3): δ (ppm) = 1.24 (m, 6H, CH2), 1.53 (m, 4H, CH2CH2CO), 2.22 (t, J = 7.5 Hz, 2H, CH2CO), 2.44 (t, J = 7.5 Hz, 2H, 8-CH2), 2.76 (t, J = 6.2 Hz, 2H, 10-CH2), 3.19 (t, J = 6.2 Hz, 2H, CH2COPh), 3.57 (s, 3H, OCH3), 7.36 (m, 2H, aromat. 3′-/5′-H), 7.47 (m, 1H, aromat. 4′-H), 7.89 (m, 2H, aromat. 2′-/6′-H).- C19H26O4 (318.42): calc. C 71.66, H 8.23; found: C 71.44, H 8.19.
2.3.2.3.3 Methyl 9,12-dioxo-12-(2'-thienyl)dodecanoate (25)
Enone 4 (0.42 g, 2 mmol) and 2-thienylcarboxaldehyde (0.29 g, 2.67 mmol) afford 73% 25 (0.47 g, 1.45 mmol). Different from the general procedure a smaller amount of the catalyst sodium cyanide (65 mg, 1.3 mmol) was used. The white colour of the product turns in a short time to a faint yellow. Rf–value: 0.29 (petroleum ether/diethyl ether = 1:1).- M.p.: 39–40 °C.
1H NMR (CDCl3): δ (ppm) = 1.29 (m, 6H, CH2), 1.59 (m, 4H, CH2CH2CO), 2.28 (t, J = 7.5 Hz, 2H, CH2CO2), 2.48 (t, J = 7.4 Hz, 2H, 8-CH2), 2.82 (t, J = 6.4 Hz, 2H, 10-CH2), 3.20 (t, J = 6.4 Hz, 2H, 11-CH2), 3.64 (s, 3H, OCH3), 7.11 (dd, J = 4.9 Hz, J = 3.8 Hz, 1H, 4′-H), 7.61 (dd, 4J = 1.1 Hz, J = 4.9 Hz, 1H, 5′-H), 7.74 (dd, 4J = 1.1 Hz, J = 3.8 Hz, 1H, 3'-H).- C17H24O4S (324.44): calc. C 62.94, H 7.46; found: C 62.83, H 7.17.
2.3.2.3.4 Methyl 9,12-dioxo-12-(2′-furyl)dodecanoate (26)
Different from the general procedure 2-furylaldehyde is added slower, namely in 90 min to the sodium cyanide solution. After stirring for 1 h enone 4 is added within 2 h and then, for further 2 h stirring is continued. Work-up is done as in the general procedure. Enone 4 (0.42 g, 2 mmol) and furfural (0.26 g, 2.67 mmol) afford 54% of 26 (0.33 g, 1.07 mmol). Rf –value: 0.23 (petroleum ether/diethyl ether = 5:3).- M.p.: 52–53 °C.- 1H NMR (CDCl3): δ (ppm) = 1.26 (m, 6H, CH2), 1.55 (m, 4H, CH2CH2CO), 2.24, 2.44 (2t, J =7.5 Hz, J = 7.4 Hz, 4H, CH2CO2, 8-CH2), 2.77, 3.08 (2t, J = 6.4 Hz, J = 6.4 Hz, 4H, 10-/11-CH2), 3.61 (s, 3H, OCH3), 6.48 (dd, J = 1.9 Hz, J = 3.4 Hz, 1H, 4′-H), 7.16 (d, J = 3.4 Hz, 1H, 3'-H), 7.53 (m, 1H, 5′-H).- C17H24O5 (308.37): calc. C 66.21, H 7.84; found: C 66.25, H 7.86.
2.3.2.3.5 Methyl 9,12-dioxo-12-(3'-pyridyl)dodecanoate (27)
Sodium cyanide (65 mg, 1.3 mmol) in DMF (2 mL) is added within 10 min at 35 °C to the solution of the aldehyde in DMF. Thereafter, the general procedure is followed. Enone 4 (0.42 g, 2 mmol) and 3-pyridylaldehyde (0.28 g, 2.67 mmol) afford 66% of 27 (0.42 g, 1.32 mmol).- Rf -value: 0.49 (petroleum ether/diethyl ether = 1:1).- M.p.: 61–62 °C.- 1H NMR (CDCl3): δ = 1.31 (m, 6H, CH2), 1.60 (m, 4H, CH2CH2CO), 2.29 (t, J = 7.5 Hz, 2H, CH2CO2), 2.51 (t, J = 7.2 Hz, 2H, 8-CH2), 2.87 (t, J = 6.0 Hz, 2H, 10-CH2), 3.26 (t, J = 6.2 Hz, 2H, 11-CH2), 3.65 (s, 3H, OCH3), 7.40 (dd, J = 4.9 Hz, J = 7.91 Hz, 1H, 5′-H), 8.23 (dt, J = 1.9 Hz, J = 7.9 Hz, 1H, 4′-H), 8.77 (dd, J = 1.5 Hz, J = 4.9 Hz, 1H, 6′-H), 9.19 (d, J = 1.9 Hz, 1H, 2'-H).- C18H25NO4 (319.40): calc. C 67.69, H 7.89, N 4.39; found: C 67.91, H 7.83, N 4.67.
2.3.2.3.6 Methyl 9-oxo-11-(1′-(2′-formyl)-pyrrolyl)-undecanoate (28)
Enone 4 (0.42 g, 2 mmol) and 1H-pyrrole-2-carboxaldehyde (29, 0.25 g, 2.67 mmol) afford 74% of 28 (0.46 g, 1.49 mmol).- Rf -value: 0.30 (petroleum ether/diethyl ether = 5:3).- M.p.: 48–49 °C.- 1H NMR (CDCl3): δ (ppm) = 1.20 (m, 6H, CH2), 1.35-1.6 (m, 4H, CH2CH2CO), 2.23, 2.27 (2t, J = 7.5 Hz, J = 7.3 Hz, 4H, CH2CO), 2.85 (t, J = 6.4 Hz, 2H, 10-CH2), 3.60 (s, 3H, OCH3), 4.47 (t, J = 6.4 Hz, 2H, 11-CH2), 6.12 (dd, J = 4.1 Hz, J = 2.6 Hz, 1H, 4′-H), 6.87 (dd, J = 1.7 Hz, J = 4.1 Hz, 1H, 3'-H), 6.98 (br. t, J = 2.6 Hz, 1H, 5′-H), 9.45 (s, 1H, HCO).- C17H25NO4 (307.39): calc. C66.42, H 8.20, N 4.56; found: C 66.57, H 8.27, N 4.63.
3 Results and discussion
3.1 Preparation of methyl 9-oxo-10-undecenoate (4)
For the allylic oxidation of terminal double bonds in 1-alkenes to 1-alkene-3-ones a number of oxidants are described. These are SeO2/tert-BuOOH 18, 19, SeO2 [S1, S = supporting information], NBS/CaCO3 [S2], CrO3/HOAc/Ac2O [S3], CrO3 · 2Pyr [S4], CrO3/dimethylpyrazole[S5], pyridinium dichromate/tert-BuOOH [S6], Na2CrO4/HOAc/Ac2O [S7], CrO3 (cat.)/tert-BuOOH [S8], Cr(CO)6 (cat.)/tert-BuOOH [S9] and Cr(VI)-montmorillonite (cat.)/tert-BuOOH [S10]. All these conversions were examined by us in small scale oxidations of methyl 10-undecenoate (2b). However, in most cases the yields were low ranging from 10 to 20%. With NBS/CaCO3 [S2] the yield was good; however, the reaction was unselective leading to a mixture of regioisomers. Very promising appeared the oxidation with Cr(VI)-montmorillonite (cat.)/tert-BuOOH [S10] and the oxidation with SeO2/tert-BuOOH 18, 19. The first reagent led to high yields of enones with terminal double bonds in shorter and unsubstituted olefins as in 1-hexene, 1-heptene and 1-decene 27. However, according to our experiments the conversion of methyl 10-undecenoate (2b) was low. In conclusion, from the large number of reagents for the allylic oxidation, only SeO2/tert-BuOOH 18, 19 proved to be useful for the C-9 oxidation of ester 2b. With 0.5 equivalents of selenium dioxide and two equivalents of tert-butyl hydroperoxide at room temperature 55% of methyl 9-hydroxy-10-undecenoate (5) and 7% of methyl 9-oxo-10-undecenoate (4) were obtained. The alcohol 5 could be oxidized with potassium dichromate in diethyl ether/sulphuric acid at 0°C to 84% of the enone 4 28. This amounts to an overall yield of 53% of the enone in a two step synthesis. Based on the conversion of 2b the yield of enone 4 is even 81%, when recovered 2b is used. Selenium dioxide can be also recycled. Additionally, the amount of selenium dioxide can be reduced to 0.1 equivalents at the cost of a slightly lower yield. Residual selenium in the product 5 is lower than 1 ppm as determined by atom absorption spectroscopy.
The enone 4 shows in the IR-spectrum an additional absorption at 1682 cm−1 for the conjugated keto group. In the mass spectrum the base peak is formed by the α-cleavage next to the keto carbonyl group (m/z = 55) and the Mc Lafferty rearrangements initiated by the keto group (m/z = 70, 83).
3.2 Reaction of CH-acidic compounds with methyl 9-oxo-10-undecenoate
3.2.1 Addition of 1,3-dicarbonyl compounds in a Michael – reaction
In the Michael – reaction nucleophiles are added to electrophilic olefins 22, 29. The addition allows introducing a large variety of functional groups into the alkene and also enables an extension of the carbon skeleton. Here we explore the Michael – reaction of 1,3-dicarbonyl compounds, nitroalkanes, hydrogen cyanide, and aromatic aldehydes with enone 4 as Michael – acceptor.
The 1,3-dicarbonyl compounds diethyl malonate (pks = 12.7) 30, ethyl acetoacetate (pKs = 10.7) 30 and acetylacetone (8) (pKs = 9) 30 are all more acidic than methanol (pKs = 16) 31. For that reason sodium methoxide is sufficiently basic to deprotonate the 1,3-dicarbonyl compounds 6, 8 and 10. This way five equivalents of the anions of 6 and 8 were added to enone 4 at RT affording the 1,5-dicarbonyl compounds 7 and 9 in 99% yield (based on 4) (Schemes (1a) and (1b)).
In the addition of diester 10 a mixture of the expected product 11 and the cyclohexenone 12 was found. Decreasing the reaction temperature to −30 °C increased the yield of 11 to 88%. On the other hand treating 11 in refluxing THF with sodium led to cyclohexenone 12 in 80% yield (Scheme (c)).

With the C18-enone 3 as Michael acceptor the addition of 6 needed 20 equivalents of 6 to achieve a 90% yield of adduct at the internal carbon atom C-10 15. With 20 equivalents of 10 and enone 3 the yield of the adduct corresponding to 11 dropped to only 2%. With 30 equivalents of 10 and four equivalents of the sodium salt of 10 an 89–96% yield of the cyclohexenone, corresponding to 12, was obtained 15. This shows that enone 4 has a significantly higher reactivity than enone 3 as Michael acceptor being probably due to a lower steric shielding of the terminal position.
The structures of 7 and 11 are supported by α-cleavages next to the keto carbonyl group (7: m/z = 185, 187 and 11: m/z = 185, 171) and the Mc Lafferty rearrangement in direction to the fatty acid ester group (7: m/z = 202, 11: m/z = 186). In 9 and 11 the α-cleavage next to the methyl carbonyl group is so much favoured that the fragment ion (m/z = 43) is the base peak. In the 1H NMR of the three compounds the three triplets of the methylene protons being in α-position to the carbonyl group indicate the retained structure of the 9-oxoundecanoyl group and the saturation of the double bond. The singlet of the methyl groups of the new introduced methoxy groups (δ = 3.6 ppm) and acetyl groups (δ = 2.09 ppm) support the corresponding additions. The 1H and 13C NMR spectra of 9 indicate a mixture of the enol and keto form. The portion of the enol form is in CDCl3 about 34%.
The IR-spectrum of 12 shows two carbonyl bands at 1739 cm−1 and at 1670 cm−1; the latter indicates that the compound contains an α,β-unsaturated carbonyl group. This is confirmed by the mass spectrum, which contains the intense fragment ions m/z = 82, 95, 110 and 123, which most probably originate by a Mc Lafferty-rearrangement of the double bond followed by the loss of carbon monoxide. The proposed structure is further supported by the singlet of the olefinic proton at 5.74 ppm in the 1H NMR spectrum.
For the formation of 12 from 11 the following reactions appear plausible: 11 undergoes an intramolecular aldol condensation between the methyl group of the acetyl group and the 9-keto group to form a cyclohexenone derivative; hydrolysis of the ester in the cyclohexenone ring followed by a decarboxylation of the ß-ketoacid leads to 12. The aldol condensation and the decarboxylation are accelerated at higher temperatures; this explains that at RT 11 is partially converted to 12, whilst at −30 °C this reaction is slower and the portion of 11 is increased. A further reduction of the temperature led to no further improvement of the selectivity. All additions are also possible with a similar yield without using a solvent.
3.2.2 Addition of nitroalkanes in a Michael – reaction
The addition of nitroalkanes to enone 4 proceeds under similar conditions as this of the 1,3-dicarbonyl compounds. Enone 4 was reacted in methanol with an excess of nitroethane, 1-nitropropane and 2-nitropropane and with sodium methoxide as base to afford 14–16 (Scheme ). The reaction of deprotonated nitromethane with enone 4, prepared from methyl nonanedioate, affording 13 has been described in ref. 21. After 3 h the enone 4 was quantitatively converted, and the corresponding methyl 12-nitro-9-oxo-10-undecenoates 14–16 were obtained in 78–89% yield as colorless oils. A multiple reaction of the acidic CH-bonds in 14 and 15 with enone 4 was not observed due to the applied excess of the nitroalkanes.

With enone 3 as Michael – acceptor and the nitroalkanes used above harsher reaction conditions were necessary to achieve similar adduct yields; these conditions were: refluxing enone 3 with 20 equivalents of the nitroalkane and four equivalents of the sodium nitroalkane 15.
The IR spectra of compounds 14–16 show the CO absorption of the ester group (1735–1780 cm −1), the keto group (1715–1725 cm−1) and the two absorptions of the nitro group at 1540–1550 cm−1 and 1350–1370 cm−1. The mass spectra display in no one of the three products the molecule ion, as this splits off NO2 leading to the fragment ions [M+-NO2]. The unchanged molecule part of 4 is indicated by the α-cleavage at the keto group (m/z = 185). In the 1H NMR spectra the methine protons (14: δ = 4.49 ppm, 15: δ = 4.35 ppm) and the signals of the terminal methyl groups identify the extended carbon frame.
The compounds 14–16 may be used for further conversions. Reduction to amines leads to 12-aminofatty acids, which could serve as monomers for polyamides. The reductive cyclization of the γ-nitroketones leads to nitrogen heterocycles as pyrrolidines 15. Of particular interest would be the Nef-reaction of the secondary nitro group, as this reaction would generate 1,4-diketones, which could be cyclized to five membered heterocycles and cyclopentenones.
3.2.3 Nef-reaction of the nitro compounds 14 and 15
Primary and secondary nitro compounds can be converted into aldehydes and ketones. This conversion, known as Nef-reaction 32 is achieved by acidifying the sodium salts of the nitro compound frequently with sulphuric acid.
The nitro compounds 14 and 15 were converted with sodium methoxide into their sodium salts and these are added to a solution of sulphuric acid in methanol at −10 °C. Using these conditions the 9,12-dioxofatty acid methyl esters 18 and 19 can be obtained quantitatively as white solids (Scheme ).

The Nef-reaction of the nitroethane adduct to enone 3 afforded the 1,4-diketone in 88% yield 15, compared to 99% of 18 from enone 4.
The structures can be unequivocally identified by the mass spectra. The two keto groups lead to α-cleavages and Mc Lafferty rearrangements, which generate the base peak and fragment ions of high intensity. Of interest are the 1H NMR spectra of the diketones 18 and 19. They contain four methylene groups being in α-position to the carbonyl groups and each should appear as triplet. Two of these appear as triplet, the other two, however, appear in 18 as multiplet and in 19 as singlet. The cause could be: the methylene protons are magnetically nearly equivalent and for this reason they experience a nearly identical chemical shift. In 19, the difference in the chemical shifts is so small that the four protons are magnetically equivalent and appear as singlet.
From the prepared 1,4-diketones the heterocycles: pyrrole, furan and thiophene should be accessible by a Paal-Knorr synthesis 33. This should lead to fatty acid derivatives with a five membered heterocyclic compound in the alkyl chain. Of particular interest is the cyclization of 1,4-diketones to cyclopentenones. This core structure is found in natural products as the rethrolons, jasmons and prostaglandins 34, which are of pharmaceutical interest. Methyl 9,12-dioxododecanoate 17 was synthesized for that purpose 21, which underlines the importance of 1,4-diketones as intermediates for the synthesis of biological active compounds.
3.2.4 Addition of methanol to methyl 9-oxo-10-undecenoate
The reaction of sodium methoxide in methanol as Michael – donor affords with enone 4 as Michael – acceptor in 86% yield the adduct methyl 11-methoxy-9-oxoundecanoate (20) (Scheme ). With enone 3 the corresponding adduct was obtained in 66% yield 15.

The IR spectrum and the mass spectrum confirm the structure of 20. The fragment ions, formed by α-cleavage (m/z = 87) and the Mc Lafferty rearrangement (m/z = 102) at the keto group are a proof for the addition of methanol. In the 1H NMR spectrum the additional triplet (δ = 3.41 ppm, J = 6.1 Hz) and a singlet (δ = 3.09 ppm) indicate the methylene and methyl protons bound to the oxygen.
The reaction indicates the possibility to add alcohols as Michael-donors and to produce fatty acid derivatives with an alkoxy group in terminal position.
3.2.5 Addition of hydrocyanic acid to methyl 9-oxo-10-undecenoate
For the conjugate addition of the nitrile group a mixture of sodium cyanide and ammonium chloride in aqueous DMF is suitable 35. The addition of the cyanide ion at 90 °C to enone 4 led to 82% of methyl 11-cyano-9-oxoundecanoate (21) as white solid (Scheme ). For hydrolysis the nitrile 21 is refluxed in conc. HCl and acetic acid 23 to afford 4-oxododecanedioic acid (22) in 92% yield. To facilitate the isolation and purification the diacid 22 is converted with methanol/2,2-dimethoxypropane into the diester 23, whose overall yield based on enone 4 is 63% (Scheme ). With enone 3 similar yields in these conversions were obtained 15. The 4-oxodiester 23 has been used frequently as intermediate in prostaglandin syntheses 24, 36.

The IR spectrum of the nitrile 21 shows the absorption of C-N triple bond at 2249 cm−1. The mass spectrum indicates the position of the keto group through the α-cleavages and the Mc Lafferty rearrangements. The 1H NMR spectrum with the methylene protons adjacent to the keto- and cyano-group is a further proof for the addition. The chemical shift of the nitrile carbon atom at δ = 118.9 ppm in the 13C NMR spectrum is an additional support of the structure.
3.2.6 Cyanide catalysed addition of aromatic and heteroaromatic aldehydes to methyl 9-oxo-10-undecenoate
The polarity of the electrophilic carbon atom in aldehydes can be changed into a nucleophile with the cyanide ion (Stetter - reaction) 26. Thereby, the cyanide anion adds to the carbonyl group, which is then followed by a proton rearrangement to a deprotonated cyanohydrin, which reacts as nucleophile in the Michael − addition to enone 4.
Catalysed by sodium cyanide in DMF, enone 4 was reacted with several aromatic and heteroaromatic aldehydes. The reaction times had to be varied dependent on the reactivity of the aldehyde. The methyl 12-phenyl-9,12-dioxododecanoate (24) and the methyl 12-heteroaryl-9,12-dioxododecanoates 25–27 were obtained in 54–73% yield (Scheme (a)). With enone 3 and benzaldehyde and pyridine-3-carbaldehyde under similar conditions adduct yields of 44% and 84%, respectively, have been obtained 15.

1H-Pyrrole-2-carboxaldehyde (29) was also reacted with enone 4. However, in this case a C-N bond was formed yielding 74% of the adduct 28 as a colorless, labile solid (Scheme (b)).
The IR-spectra of the compounds 24–27 show three carbonyl bands, whose wave number is characteristic for the ester group, the aliphatic and the α,β-unsaturated keto group. The mass spectra are characterized by α-cleavages and Mc Lafferty-rearrangements of the carbonyl groups. In the 1H NMR spectra new signals appear in the region for aromatic compounds and the four triplets of the methylene protons groups being in α-position to the carbonyl groups.
In the 1H NMR spectrum of 28 the triplet of the 11-methylene protons (δ = 4.47 ppm, J = 6.4 Hz) is shifted downfield. This cannot be explained by an adjacent carbonyl group but points to a bond to a nitrogen atom. The singlet of the aldehyde proton at 9.45 ppm proves the presence of the heteroaromatic formyl group in the product. In the 13C NMR the multiplicity and position of the aldehyde carbon atom (δ = 178.9 ppm (d)) is a further proof of the structure.
The synthesis of methyl ω-hetaryl-9,12-dioxododecanoates 25–27 complements the preparation of fatty acids with internal heteroaromatic groups being accessible by Paal-Knorr synthesis from 1,4-diketones 15. Stetter has extended the Michael – addition to aliphatic aldehydes using thiazolium salts as catalysts, whose basicity is sufficiently low that competing aldol reactions are excluded 37. However, the application of a thiazolium salt in the addition of propanal to enone 3 led to only 4% of adduct 15.
The C-N bond formation with the pyrrole 29 indicates the possibility to use amines as Michael donors and this way develop a way to N-substituted fatty acids being an alternative to the reduction of the nitro- or nitrile group.
4 Conclusion
Methyl 10-undecenoate (2b) is accessible by thermal cleavage of ricinoleic acid (1a), which is obtained from castor oil. The attempted allylic oxidation of 2b with many reagents, reported in the literature for this purpose, proceeded unsatisfactorily due to low conversions and unselective reactions. With 0.5 equivalents of selenium dioxide and two equivalents of tert-butyl hydroperoxide 18, 19 the ester 2b is oxidized to a mixture of methyl 9-oxo-10-undecenoate (4) and methyl 9-hydroxy-10-undecenoate (5) in 62% yield; ester 5 can be oxidized with potassium dichromate in 84% yield to ester 4. In this two step oxidation 53% of 4 are obtained; the yield can be increased to 81% of 4 by including the oxidation of recovered 2b.
The electrophilic double bond of enone 4 enables numerous nucleophilic additions. 1,3-Dicarbonyl compounds can be added to enone 4 in good to excellent yield to afford substituted ketoesters and triesters. The Michael-addition of nitroalkanes provides methyl 12-nitro-tri- and tetradecanoates in high yield. The secondary nitro compounds can be converted by the Nef- reaction in 99% yield into 1,4-dicarbonyl compounds that are precursors for five membered heterocycles 15. By addition of hydrogen cyanide, hydrolysis of the nitrile and esterification of the acid the diester 23 is accessible in 63% yield from enone 4. The use of 23 for the preparation of prostaglandin PGB1 indicates the importance of the compound as intermediate for the synthesis of natural products. By “umpolung” with the cyanide anion in a Stetter – reaction aryl- and hetaryl aldehydes are converted to ω-aryl- and ω-hetaryl-substituted 1,4-diketones.
The yields in these reactions are higher than these obtained in corresponding additions at the carbon atom C-10 of enone 3 obtained from methyl ricinoleate. This indicates a higher reactivity of the terminal enone 4 compared to the internal enone 3. A reason could be a stronger steric shielding of the disubstituted internal double bond compared to the monosubstituted terminal one. Thus the additions to enone 4 not only lead to higher yields but also provide with the terminal addition an alternative to the internal nucleophilic addition with enone 3.
The accessible compounds should be useful as building blocks for polymers, pharmaceuticals and as components in fatty acid conjugates for the attachment of bioactive components and of groups with suitable physical properties for the design of materials.
Support of this work by the Minister of Research and Technology of FRG (Projekt: Intensivierung der Forschung auf dem Gebiet der Fettchemie) is gratefully acknowledged.
The authors have declared no conflict of interest.

