
Molecular Genetics & Genomic Medicine

Journal list menu

 Issue
IssueMolecular Genetics & Genomic Medicine: Volume 2, Issue 6
i-iii, 455-547November 2014
Export Citations
Download PDFs
Issue Information
Invited Commentaries
Driving personalized medicine forward: the who, what, when, and how of educating the health-care workforce
- Pages: 455-457
- First Published: 14 September 2014
Medical Genetics in Paraguay
- Pages: 458-466
- First Published: 03 November 2014
Medical genetics in Paraguay is still in its infancy, with many tests being sent out of the country given the lack of local availability, no genetic counselors, no accredited post-doctoral residency training program in genetics, and only one clinical geneticist serving a country of almost 7 million people.
Original Articles
Terminal osseous dysplasia with pigmentary defects (TODPD) due to a recurrent filamin A (FLNA) mutation
- Pages: 467-471
- First Published: 08 August 2014
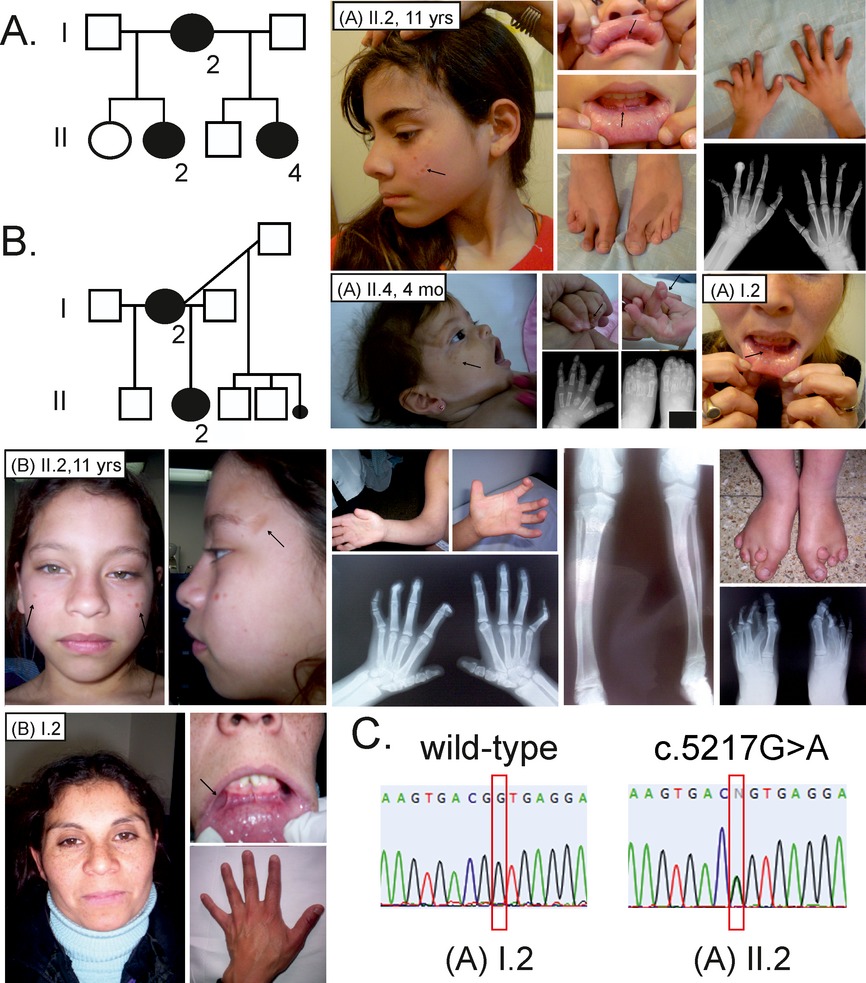
Terminal osseous dysplasia with pigmentary defects (TODPD) is an X-linked dominant syndrome with distal limb anomalies, pigmentary skin defects, digital fibromas, and generalized bone involvement due to a recurrent mutation in the filamin A (FLNA) gene. We here report the mutation c.5217G>A in FLNA in three families with TODPD and we found possible germline and somatic mosaicism in two out of the three families.
Exome analysis identifies Brody myopathy in a family diagnosed with malignant hyperthermia susceptibility
- Pages: 472-483
- First Published: 06 June 2014
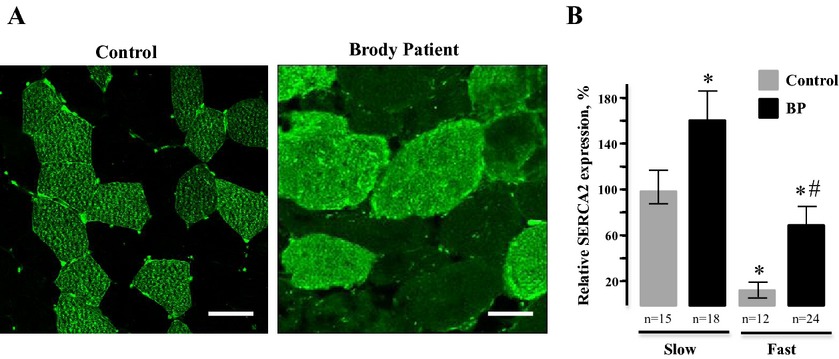
We identified and characterized Brody myopathy in a family diagnosed with malignant hyperthermia (MH). Exome sequencing revealed compound heterozygous mutations in the SERCA1, a calcium pump, expressed in fast-twitch muscles. Analyses of affected muscles show the absence of SERCA1, but upregulation of SERCA2 demonstrating a compensatory mechanism that partially restores the diminished Ca2+ transport. MH diagnosis in the family is likely due to prolonged elevation of intracellular Ca2+.
Functional and structural impact of the most prevalent missense mutations in classic galactosemia
- Pages: 484-496
- First Published: 23 June 2014
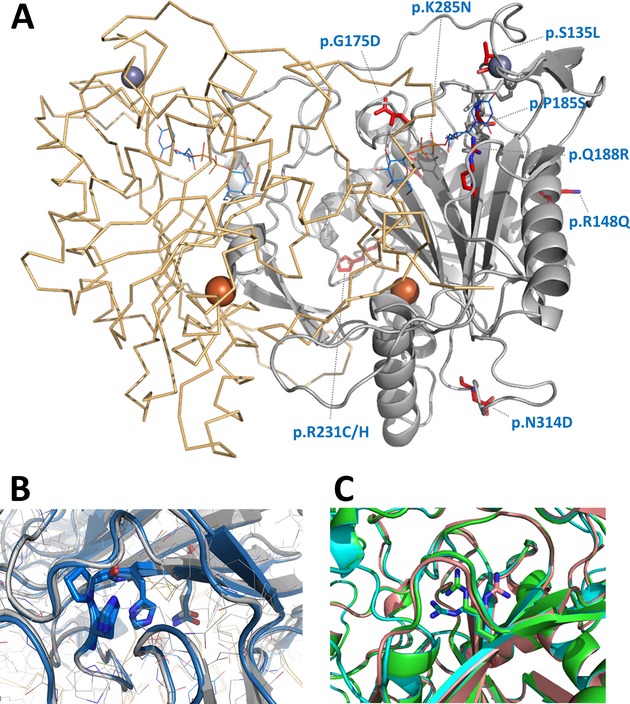
Classic galactosemia results from deficiency in galactose-1-phosphate urydylyltransferase, GALT, a key enzyme in galactose metabolism. Herein, we show that the most frequent GALT mutations result in disturbed protein aggregation despite limited effects on secondary and tertiary structures. These observations shed new light on the molecular basis for pathogenicity in the most frequent GALT mutations, suggesting alternative therapeutic interventions for classic galactosemia.
FGFR3 mutation frequency in 324 cases from the International Skeletal Dysplasia Registry
- Pages: 497-503
- First Published: 05 August 2014
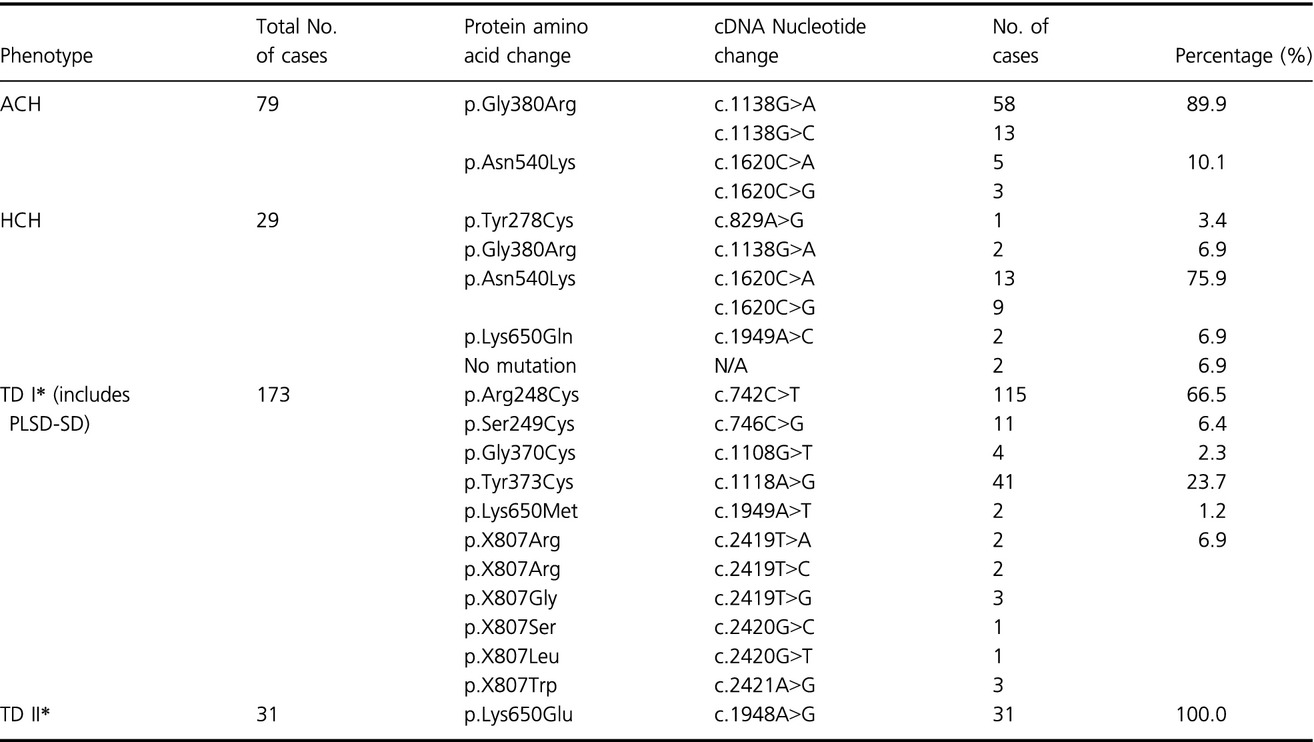
Based on the largest number of skeletal dysplasia cases, this study provides valuable information on Fibroblast growth factor receptor 3 (FGFR3) mutation frequency of four skeletal dysplasias for clinical diagnostic laboratories and clinicians.
Ten new ATM alterations in Polish patients with ataxia-telangiectasia
- Pages: 504-511
- First Published: 30 July 2014
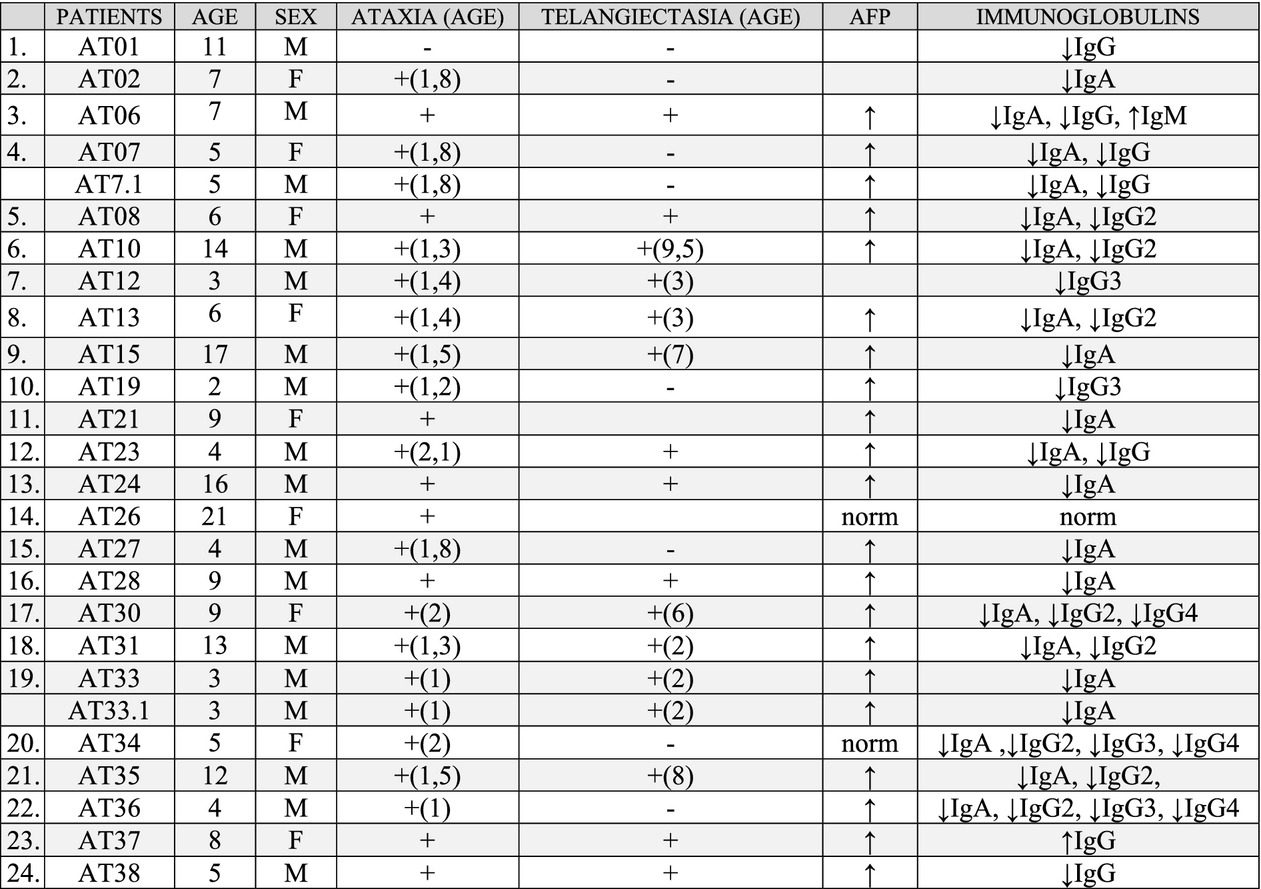
The objective of the present study was molecular analysis of ATM gene in a cohort of 24 Polish patients with ataxia-telangiectasia in aim to provide an updated mutational spectrum in Polish AT patients. As result of molecular analysis status of recurrent mutation was confirmed and also ten new ATM variants were detected.
A common cognitive, psychiatric, and dysmorphic phenotype in carriers of NRXN1 deletion
- Pages: 512-521
- First Published: 18 August 2014
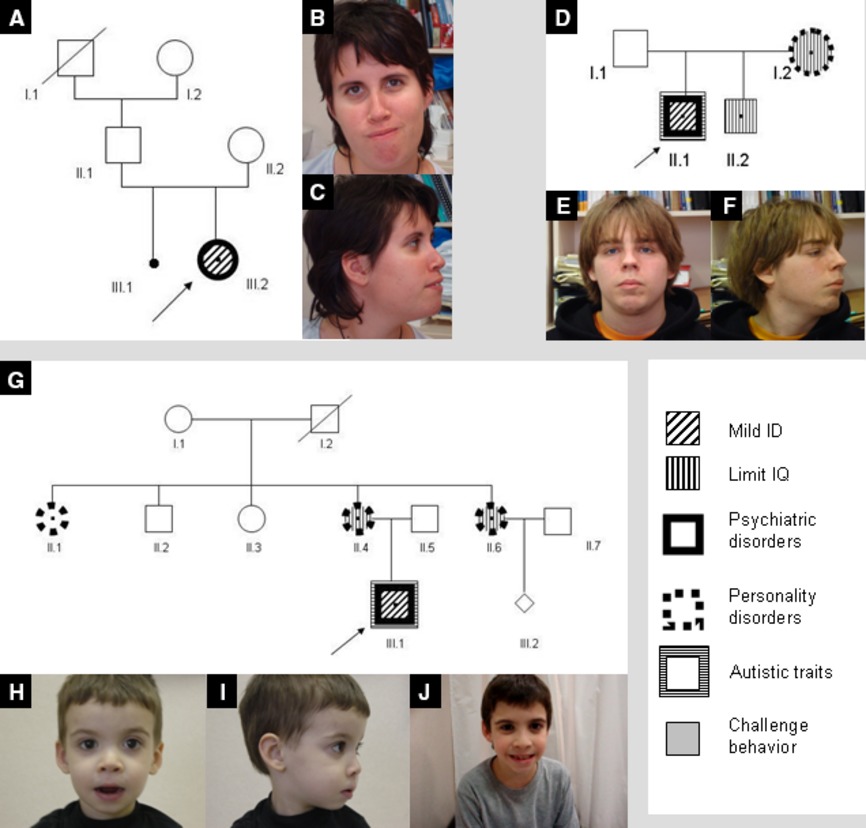
Array comparative genomic hybridization identifies a 2p16.3 deletion that includes the NRXN1 gene in three unrelated patients who share the same dysmorphic, cognitive, and psychiatric phenotype. The exhaustive neuropsychological assessment of the family members who carry the deletion showed an intelligence quotient from borderline to average and all present an anxiety disorder and a dysexecutive syndrome. We suggest that the NRXN1 gene is a risk factor for intellectual disability and psychiatric disorder with variable expressivity.
The allelic spectrum of Charcot–Marie–Tooth disease in over 17,000 individuals with neuropathy
- Pages: 522-529
- First Published: 21 August 2014
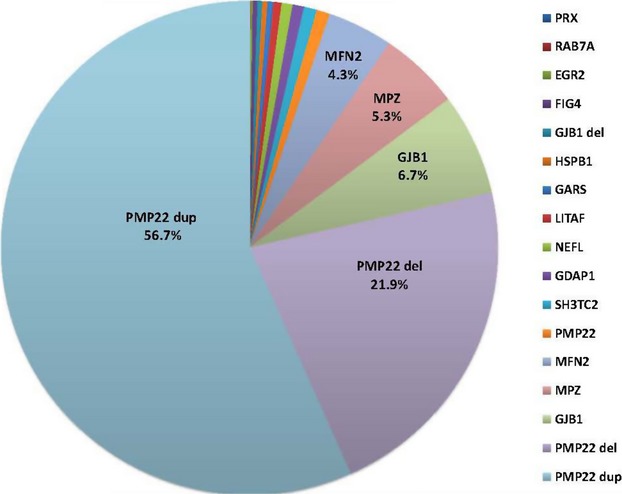
In a testing cohort of 17,880 individuals, we report that duplications (dup) (56.7%) or deletions (del) (21.9%) in the PMP22 gene accounted for the majority of positive findings followed by mutations in the GJB1 (6.7%), MPZ (5.3%), and MFN2 (4.3%) genes.
Diagnosis of an imprinted-gene syndrome by a novel bioinformatics analysis of whole-genome sequences from a family trio
- Pages: 530-538
- First Published: 26 August 2014
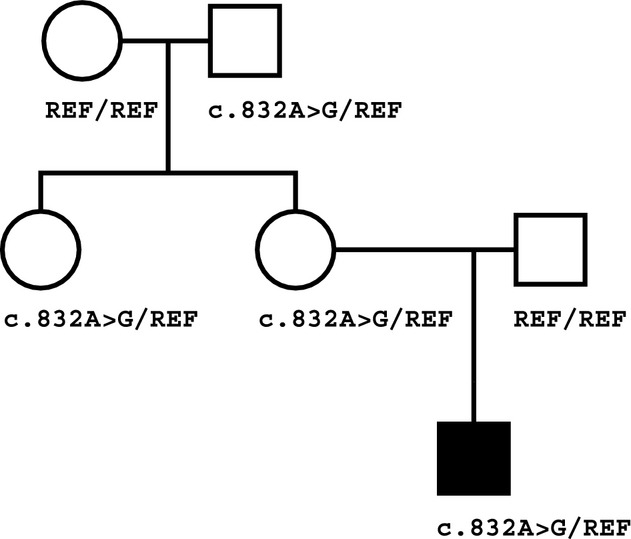
We describe a unique bioinformatic analysis of whole-genome sequences from a family trio designed to identify imprinted gene mutations that are often overlooked by inheritance-based analyses. Application of this approach to whole-genome sequencing data from a family with an affected child for whom multiple lines of investigation were nonexplanatory identified a CDKN1C mutation, thereby providing a diagnosis of IMAGe syndrome for the proband.
Complex genomic rearrangements in the dystrophin gene due to replication-based mechanisms
- Pages: 539-547
- First Published: 15 September 2014
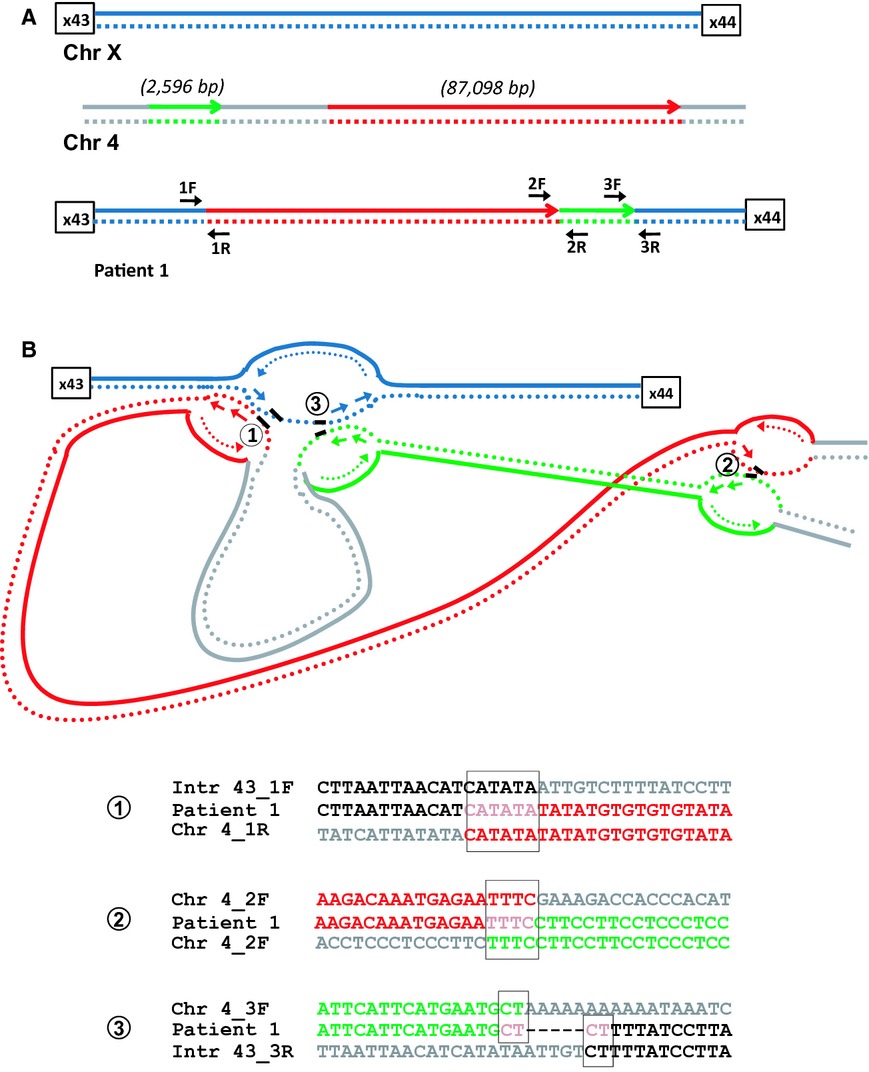
Five Duchenne muscular dystrophy (DMD) patients with complex genomic rearrangements were investigated using a combination of MLPA/mRNA transcript analysis/custom array CGH and breakpoint sequence analysis to investigate the mechanisms for these rearrangements. We propose replication-based mechanisms for all five complex DMD rearrangements.




