
Molecular Genetics & Genomic Medicine

Journal list menu

 Issue
IssueMolecular Genetics & Genomic Medicine: Volume 2, Issue 5
i-iii, 363-453September 2014
Export Citations
Download PDFs
Issue Information
Invited Commentaries
The evolution of comparative genomics
- Pages: 363-368
- First Published: 05 September 2014
The field of comparative genomics arose hand-in-hand with the ability to generate genomic sequence data. In this article I highlight some prominent applications of comparative genomic methods and describe some of the computational challenges the field encountered.
Genetics and genomic medicine in Saudi Arabia
- Pages: 369-378
- First Published: 30 July 2014
Clinical Report
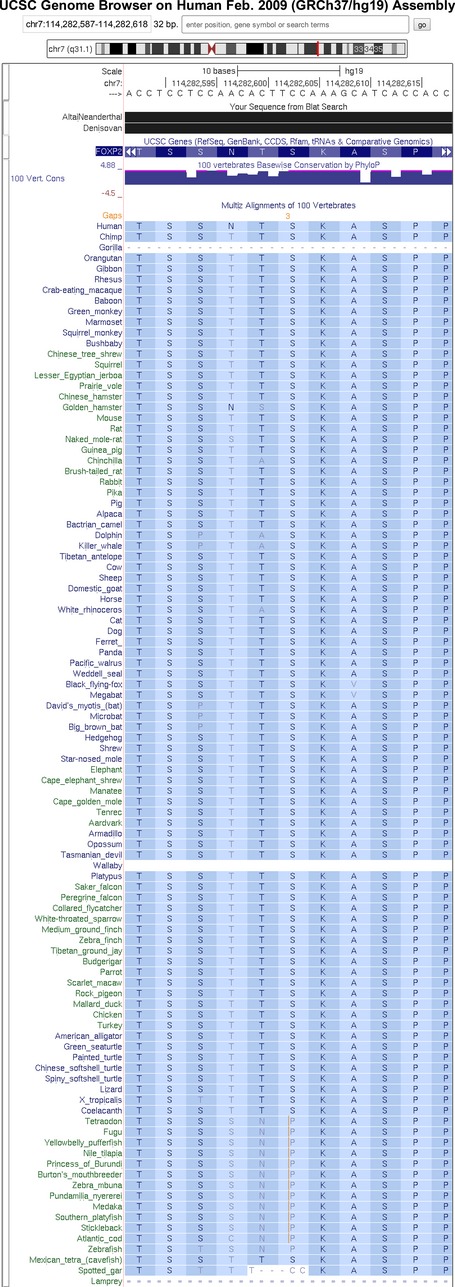
AP5Z1/SPG48 frequency in autosomal recessive and sporadic spastic paraplegia
- Pages: 379-382
- First Published: 25 May 2014
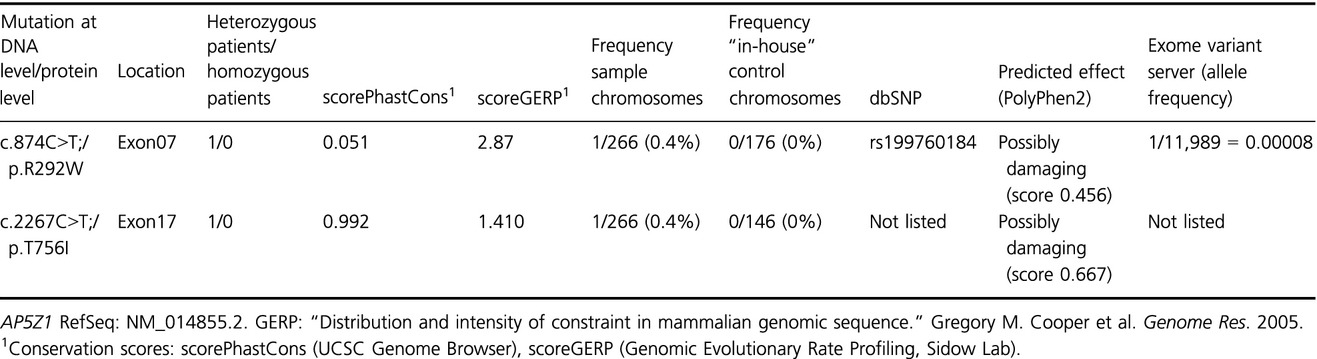
Here, we established amplicon-based high-throughput genotyping for autosomal recessive HSP. We determined the frequency of SPG48 in a large group of 127 autosomal recessive and sporadic spastic paraplegia (ARHSP) patients. Given that, up to now concerning the number of examined patients, we carried out the largest study for SPG48 and we could not identify disease-causing mutations by neither mutation scanning nor gene dosage analysis; we consider SPG48 as being rare.
Methods
Selected reaction monitoring as an effective method for reliable quantification of disease-associated proteins in maple syrup urine disease
- Pages: 383-392
- First Published: 04 June 2014
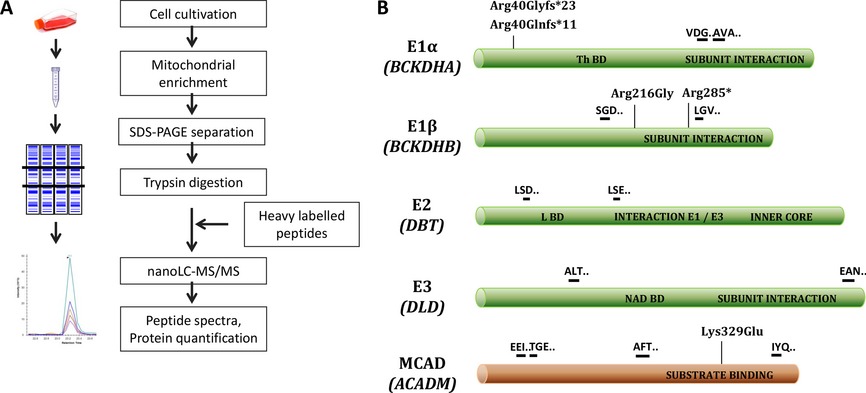
Using selected reaction monitoring (SRM) we elucidated the protein effects of mutations generating premature termination codons or misfolded proteins in fibroblasts from patients with maple syrup urine disease (MSUD). MSUD is caused by the defective activity of the branched-chain α-ketoacid dehydrogenase (BCKDH), a mitochondrial multienzyme complex encoded by four different genes. All four proteins were successfully quantified in controls, whereas E1α and E1β proteins were not detected in patients carrying mutations in one of those genes, indicating instability of E1α and E1β monomers.
Screening for single nucleotide variants, small indels and exon deletions with a next-generation sequencing based gene panel approach for Usher syndrome
- Pages: 393-401
- First Published: 15 June 2014
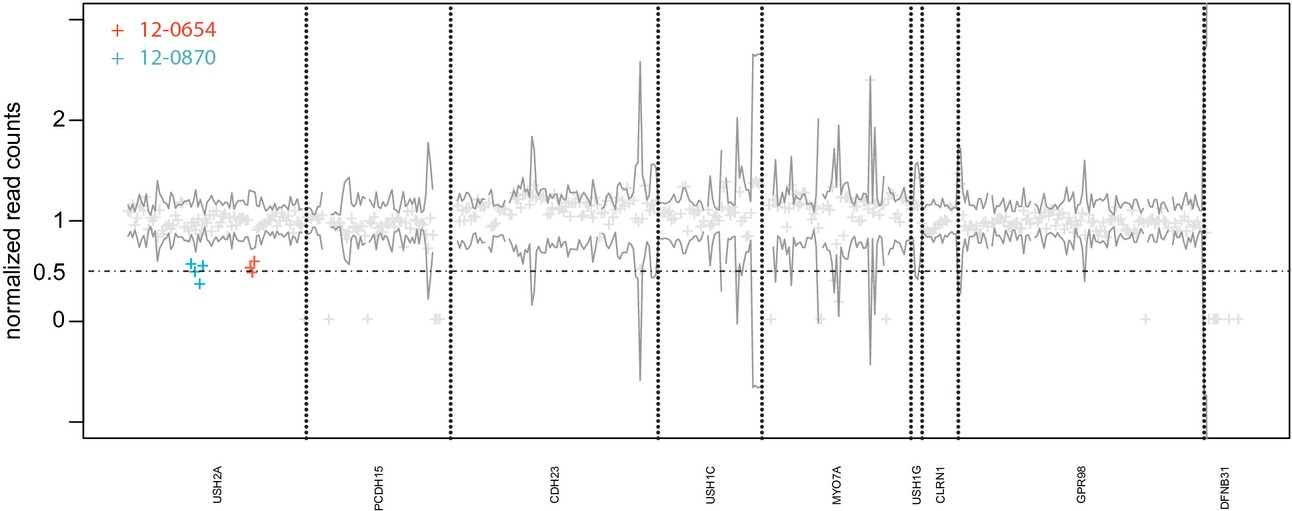
We present a comprehensive diagnostic approach for Usher syndrome that allows to detect not only single-nucleotide variants and short indels but also exon deletions in a single test. In a cohort of 44 individuals with Usher syndrome, we achieved a high diagnostic yield and report 39 new pathogenic mutations.
Original Articles
Identification of three novel FGF16 mutations in X-linked recessive fusion of the fourth and fifth metacarpals and possible correlation with heart disease
- Pages: 402-411
- First Published: 14 May 2014
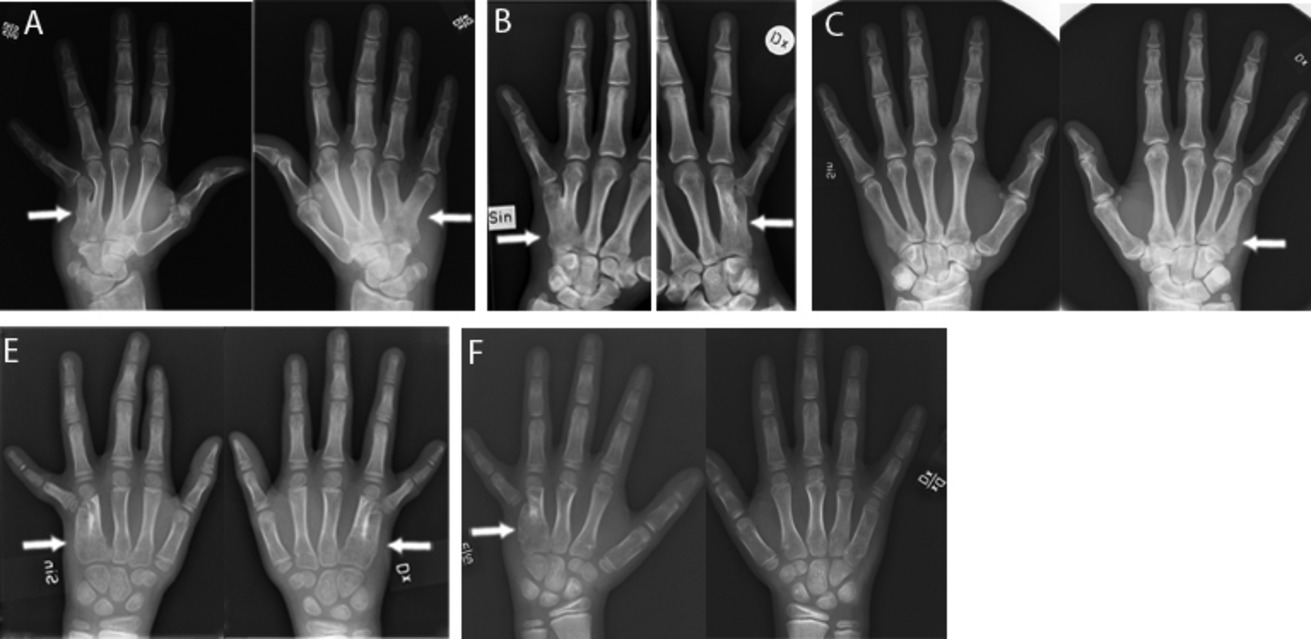
Nonsense mutations in FGF16 have recently been linked to X-linked recessive hand malformations with fusion between the fourth and the fifth metacarpals and hypoplasia of the fifth digit (MF4; MIM#309630). The purpose of this study was to perform careful clinical phenotyping and to define molecular mechanisms behind X-linked recessive MF4 in three unrelated families. We performed whole-exome sequencing, and identified three novel mutations in FGF16. The functional impact of the identified FGF16 changes were confirmed using mutated human FGF16 message and morpholino-based suppression of Fgf16 in zebrafish. In addition, clinical investigations revealed reduced penetrance and variable expressivity of the MF4 phenotype. Cardiac disorders, including myocardial infarction and atrial fibrillation followed the X-linked FGF16 mutated trait in one large family. Our findings establish that a mutation in exon 1, 2 or 3 of FGF16 results in X-linked recessive MF4 and expand the phenotypic spectrum of FGF16 mutations to include a possible correlation with heart disease.
Diagnosis of autosomal dominant polycystic kidney disease using efficient PKD1 and PKD2 targeted next-generation sequencing
- Pages: 412-421
- First Published: 23 May 2014
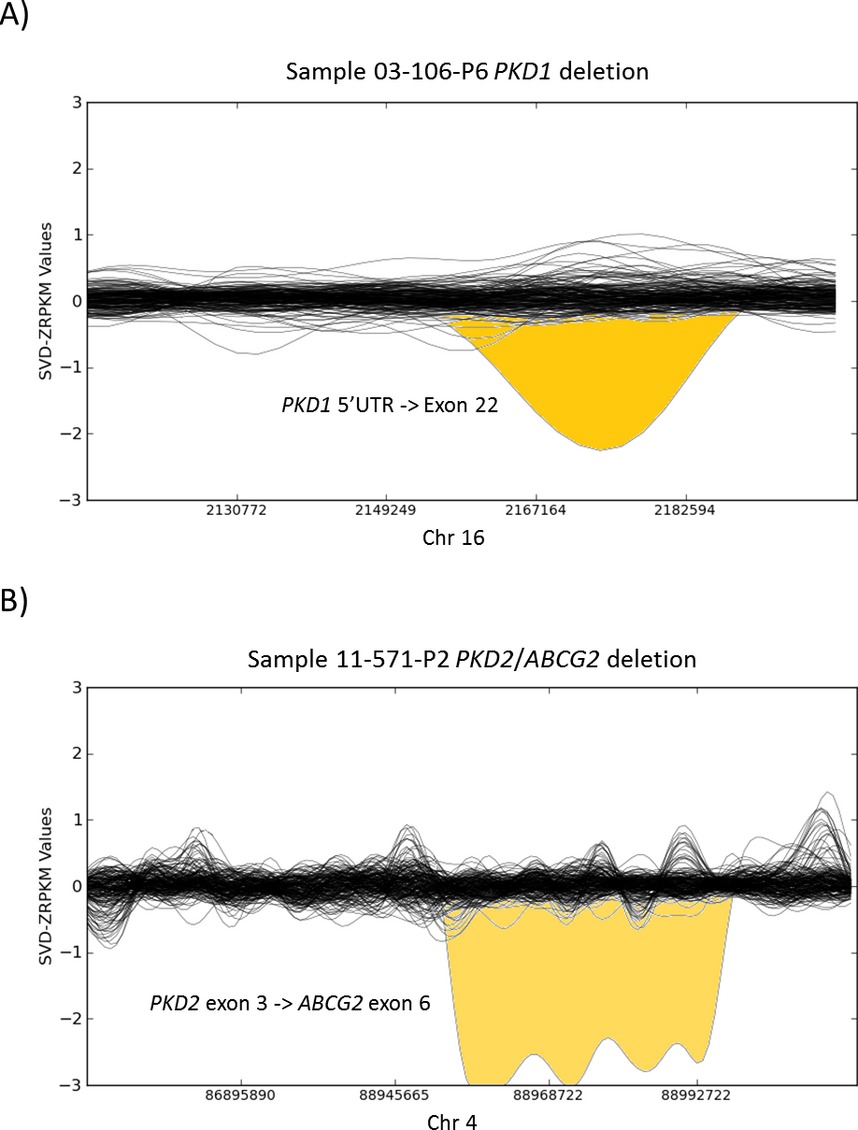
Autosomal dominant polycystic kidney disease (ADPKD) is caused by mutations in PKD1 and PKD2. To date, genetic diagnosis of ADPKD by conventional techniques requires cumbersome long-range polymerase chain reaction (PCR) of the repeated region of PKD1 followed by nested PCRs. For the first time, we successfully applied in-solution hybridization to capture the complex PKD1 gene, which is duplicated six times resulting in six pseudogenes sharing 97.7% sequence identity with the genuine gene, in addition to the PKD2 gene. Our sophisticated bioinformatics analysis was able to detect causal single-nucleotide variants as well as to characterize small insertions/deletions and large structural variants, even in the repeated region of PKD1.
Craniofacial morphometric analysis of individuals with X-linked hypohidrotic ectodermal dysplasia
- Pages: 422-429
- First Published: 20 May 2014
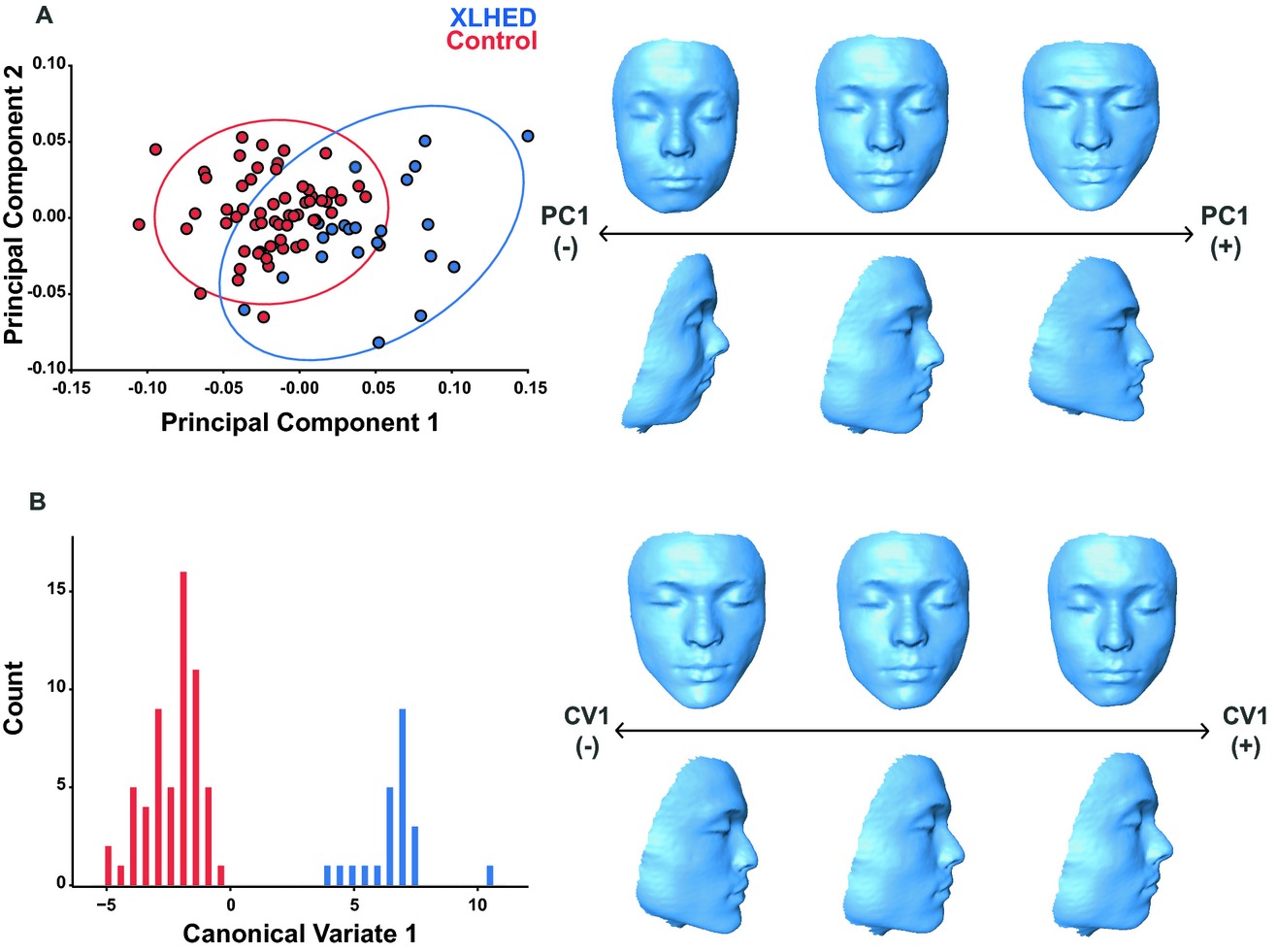
X-linked hypohidrotic ectodermal dysplasia (XLHED) is characterized by missing or malformed ectodermal structures, including skin, hair, sweat glands, and teeth. We quantitatively analyzed the XLHED craniofacial phenotype using 3D imaging and geometric morphometrics (GM) and found that individuals with XLHED have a distinct craniofacial phenotype that differed significantly from controls. Our findings extend the phenotype of XLHED and may be useful for both clinical diagnosis and treatment of XLHED.
Disease-related mutations among Caribbean Hispanics with familial dementia
- Pages: 430-437
- First Published: 04 June 2014
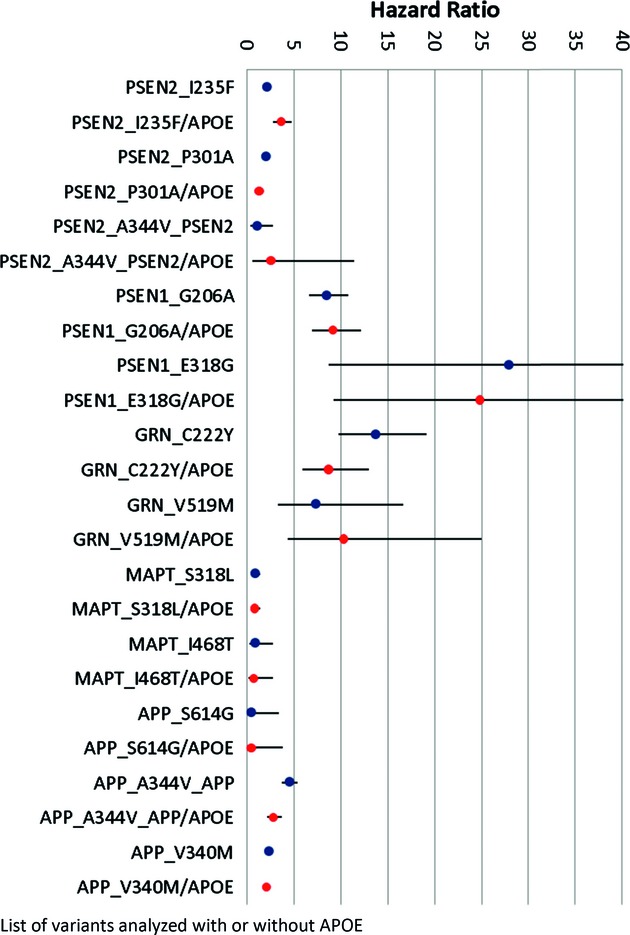
Resquencing of five genes (PSEN1, PSEN2, APP, GRN, and MAPT) in Caribbean Hispanics with familial AD identified potentially pathogenic variants in ~29.2%, where four were novel mutations, while eight had been previously observed. In addition, some family members carried variants in the GRN and MAPT genes which are associated with frontotemporal lobar degeneration.
Disease variants in genomes of 44 centenarians
- Pages: 438-450
- First Published: 15 June 2014
In genome sequences of 44 Ashkenazi centenarians, we identified many coding variants that were annotated as “pathogenic” or “likely pathogenic” based on the ClinVar database. Our data demonstrate that the incidental finding of certain reported disease variants in an individual genome may not preclude an extraordinarily long life. When the observed variants are encountered in the context of clinical sequencing, it is thus important to exercise caution in justifying clinical decisions.
Alport syndrome caused by a COL4A5 deletion and exonization of an adjacent AluY
- Pages: 451-453
- First Published: 28 May 2014
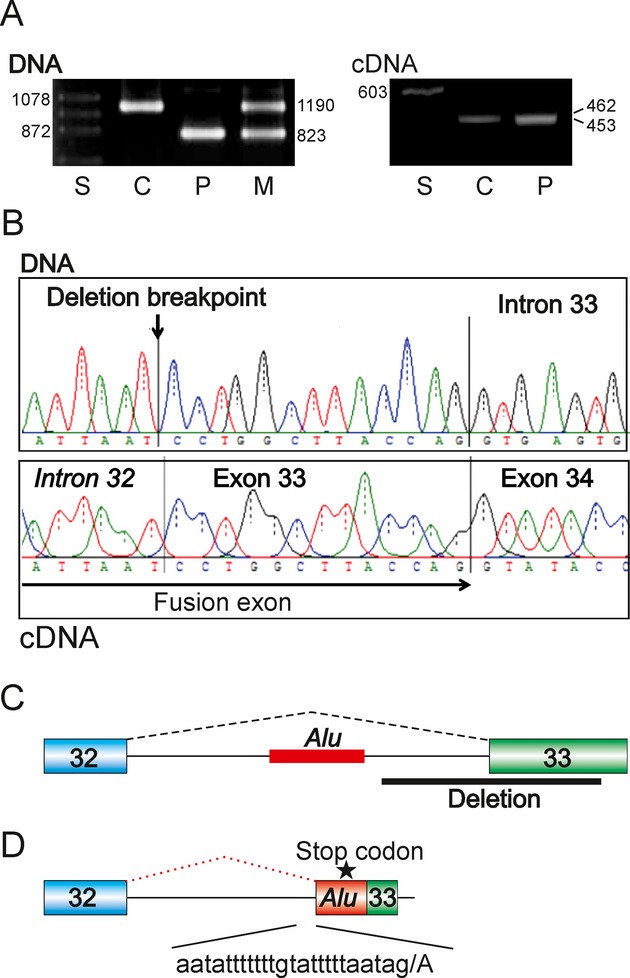
We describe a novel mechanism of deletion-induced Alu exonization, illustrating that deletions outside any transposed elements can activate splice sites within Alus and lead to new exon creation and genetic disease. Description of such events is underreported and is most useful for understanding how splice-site sequences are recognized by the spliceosome machinery, as exemplified by a recent publication in Cell 152, 453–466, 2013.




