

Journal list menu

 Issue
IssueClinical Genetics: Volume 94, Issue 3-4
281-398October 2018
Export Citations
Download PDFs
ISSUE INFORMATION
INVITED REVIEW
Unraveling molecular pathways shared by Kabuki and Kabuki-like syndromes
- Pages: 283-295
- First Published: 31 January 2017

ORIGINAL ARTICLES
Genome-wide compound heterozygosity analysis highlighted 4 novel susceptibility loci for congenital heart disease in Chinese population
- Pages: 296-302
- First Published: 17 May 2018

A ZPR1 mutation is associated with a novel syndrome of growth restriction, distinct craniofacial features, alopecia, and hypoplastic kidneys
- Pages: 303-312
- First Published: 30 May 2018


Enrichment of rare copy number variation in children with developmental language disorder
- Pages: 313-320
- First Published: 30 May 2018

How do consent forms for diagnostic high-throughput sequencing address unsolicited and secondary findings? A content analysis
- Pages: 321-329
- First Published: 10 June 2018

Graphical representation of the categories and subcategories identified in the forms. †Value includes 13 forms that did not mention unsolicited findings (UF) and/or secondary findings (SF).
Genotype-phenotype correlations of low-frequency variants in the complement system in renal disease and age-related macular degeneration
- Pages: 330-338
- First Published: 11 June 2018

The GBA p.Trp378Gly mutation is a probable French-Canadian founder mutation causing Gaucher disease and synucleinopathies
- Pages: 339-345
- First Published: 19 June 2018
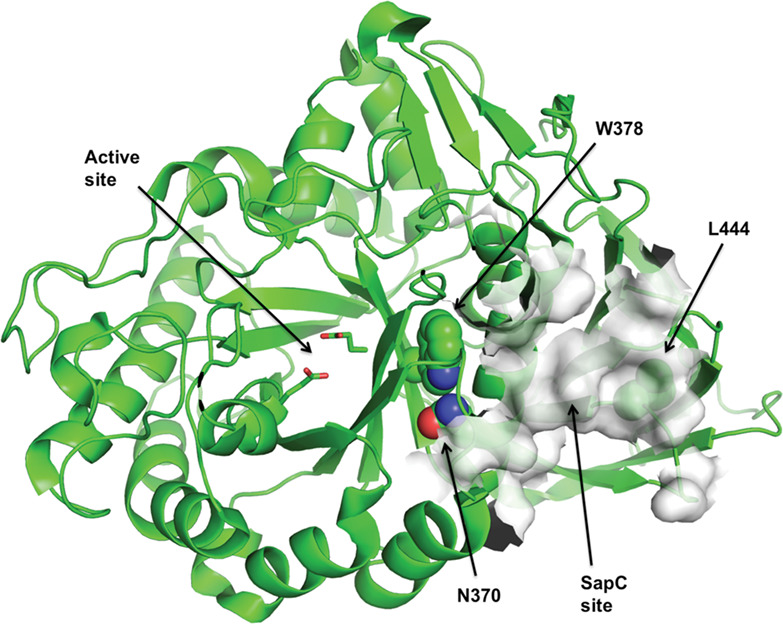
A French-Canadian founder mutation in GBA, p.Trp378Gly, is located between the enzyme active site and the binding area of Saposin C. This mutation causes Gaucher disease in compound heterozygous carriers, with or without Parkinson disease, and is a risk factor for synucleinopathies in heterozygous carriers.
SHORT REPORTS
Factors associated with ATXN2 CAG/CAA repeat intergenerational instability in Spinocerebellar ataxia type 2
- Pages: 346-350
- First Published: 14 May 2018

PPP1R21 homozygous null variants associated with developmental delay, muscle weakness, distinctive facial features, and brain abnormalities
- Pages: 351-355
- First Published: 28 May 2018

GPT2 mutations cause developmental encephalopathy with microcephaly and features of complicated hereditary spastic paraplegia
- Pages: 356-361
- First Published: 07 June 2018

PRUNE1-related disorder: Expanding the clinical spectrum
- Pages: 362-367
- First Published: 24 May 2018
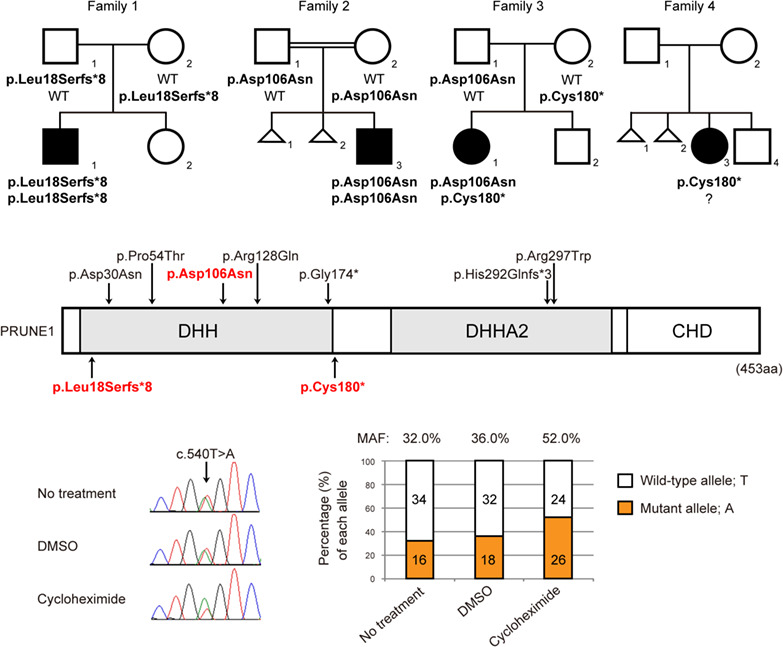
Neurodevelopmental disorder with microcephaly, hypotonia, and variable brain anomalies (NMIHBA) (OMIM #617481) is an autosomal recessive disease characterized by progressive microcephaly, plagiocephaly, hypotonia, spastic quadriparesis, global developmental delay, intellectual disability, optic features and abnormal brain magnetic resonance imaging (MRI). NMIHBA was recently reported to be caused by PRUNE1 mutations. Here, we report 3 PRUNE1 mutations in 1 Caucasian and 3 Japanese families. The patients presented in this study showed atypical NMIHBA phenotypes with no progressive microcephaly. Furthermore, one Caucasian case had significant macrocephaly; therefore, patients with PRUNE1 mutations can exhibit a broad and heterogeneous spectrum of phenotypes.
IFT80 mutations cause a novel complex ciliopathy phenotype with retinal degeneration
- Pages: 368-372
- First Published: 20 June 2018
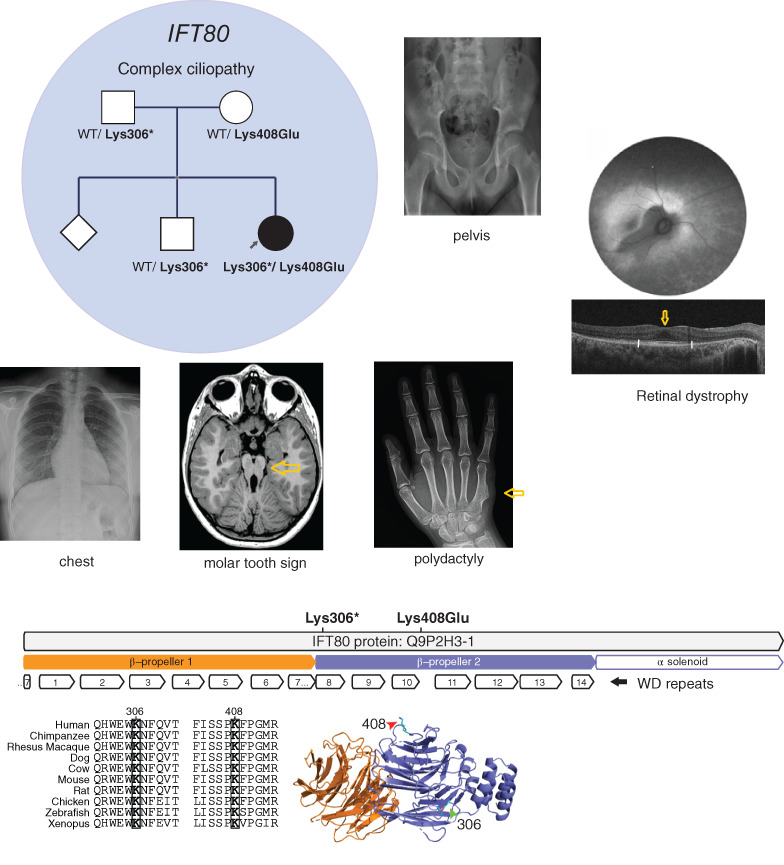
Novel IFT80 mutations (Lys408Glu and Lys306*) were identified in a patient with none of the features of Jeune syndrome (normal chest and pelvic x-rays) but retinal degeneration and a phenotype otherwise reminiscent of OFD6 (molar tooth sight, brain hamartoma digit anomaly). The amino acids involved were highly conserved and predicted to lead to the loss of most on the C-terminal domain on protein modeling. This expands the phenotype spectrum associated with IFT80 mutations.
IL11RA-related Crouzon-like autosomal recessive craniosynostosis in 10 new patients: Resemblances and differences
- Pages: 373-380
- First Published: 21 June 2018
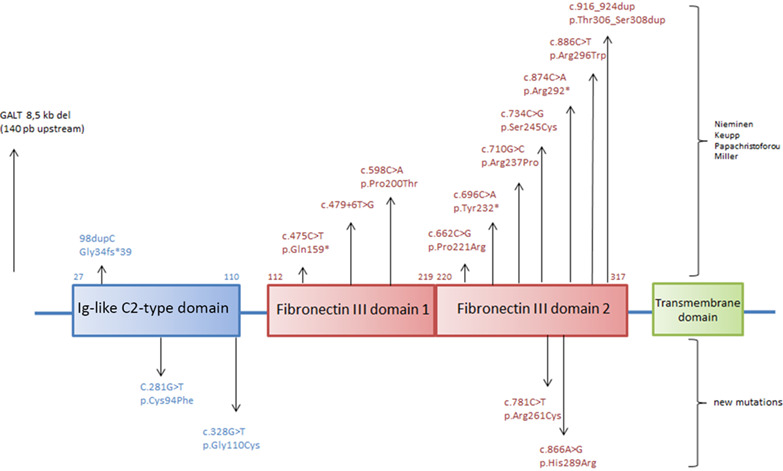
By describing 10 new patients recruited in centers for Human Genetics, we further delineate the clinical spectrum of a Crouzon-like craniosynostosis disorder, officially termed craniosynostosis and dental anomalies (MIM614188). Singularly, it is inherited according to an autosomal recessive mode of inheritance. We identified six missense mutations in IL11RA, a gene encoding the alpha subunit of interleukin 11 receptor, 4 of them being novel, including 2 in the Ig-like C2-type domain.
Evidence for HNRNPH1 being another gene for Bain type syndromic mental retardation
- Pages: 381-385
- First Published: 25 June 2018

LETTERS TO THE EDITOR
Phenotypic variability in xeroderma pigmentosum group G: An uncommon case with severe prenatal-onset Cockayne syndrome features
- Pages: 386-388
- First Published: 11 May 2018

Mutational analysis of CCM1, CCM2 and CCM3 in a Han Chinese cohort with multiple cerebral cavernous malformations in Taiwan
- Pages: 389-390
- First Published: 22 May 2018

Bilateral cerebellar cysts and cerebral white matter lesions with cortical dysgenesis: Expanding the phenotype of LAMB1 gene mutations
- Pages: 391-392
- First Published: 10 June 2018

LAMB1 gene analysis should be considered for intellectually disabled patients with cerebellar cysts, white matter signal change, and cortical malformation. Muscular involvement is absent, in contrast to the α-dystroglycanopathy types of congenital muscular dystrophies.
A founder nonsense variant in NUDT2 causes a recessive neurodevelopmental disorder in Saudi Arab children
- Pages: 393-395
- First Published: 30 July 2018
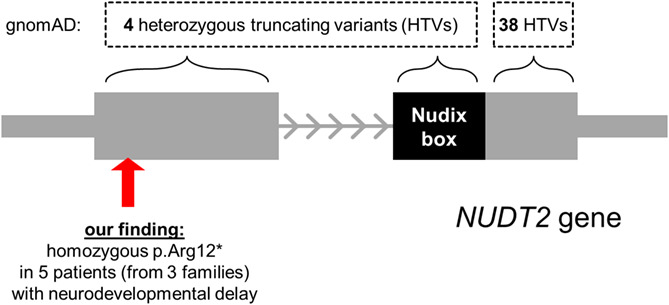
We identified the homozygous p.Arg12* variant in 5 patients with neurodevelopmental delay, but variation databases list many truncating heterozygous variants for this small 2-exon gene. As most of these affect the protein’s C-terminus, loss-of-function mediated pathogenicity may be confined to bi-allelic truncating variants in exon 1 (nonsense-mediated decay!) or in the catalytically active Nudix box.
A novel homozygous frame-shift variant in the LHCGR gene is associated with primary ovarian insufficiency in a Pakistani family
- Pages: 396-397
- First Published: 17 July 2018
