

Journal list menu

 Issue20161-134
Issue20161-134
Export Citations
Download PDFs
Cover Image, Volume 37, Issue 1
- First Published: 15 December 2015

On the cover: A heterozygous genetic lesion in a family suffering from a connective tissue disorder results in the p.A22T missense mutation in the signal peptide cleavage site of the extracellular matrix protein EMILIN1. This mutation causes a reduction in extracellular matrix deposition, an increased cellular apoptosis, and an abnormal accumulation of EMILIN-1 in the endoplasmic reticulum. Top, immunofluorescence staining of EMILIN-1 (green) in normal (left) and proband (right) skin biopsies; bottom, normal (left) and proband (right) dermal fibroblasts. See Capuano et al., page 84 in this issue.
The Genome Editing Revolution in Livestock Marches on
- Page: 5
- First Published: 15 December 2015
Mutation Update for COL2A1 Gene Variants Associated with Type II Collagenopathies
- Pages: 7-15
- First Published: 07 October 2015
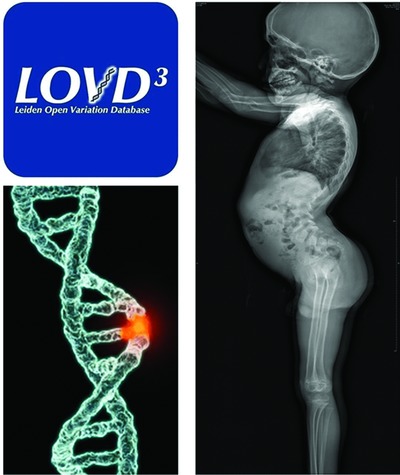
Type II collagenopathies cause a spectrum of rare autosomal dominant conditions characterized by skeletal dysplasia, short stature, and sensorial defects. A differential diagnosis of with other genetic causes of bone dysplasia is sometimes challenging. There are no recent exhaustive descriptions of mutations in the COL2A1 gene. We provide a review of COL2A1 mutations extracted from the Leiden Open Variation Database that we updated with data from PubMed and our own patients. Over 700 patients were recorded, harboring 415 different mutations.
Mutation Update of ARSA and PSAP Genes Causing Metachromatic Leukodystrophy
- Pages: 16-27
- First Published: 14 October 2015

- MLD is caused by variants in the ARSA gene or, more rarely, in the PSAP gene. An extensive review of the 200 ARSA and 10 PSAP causative variants, described in a total of 458 MLD patients and published in the literature to date is here presented, and uploaded on the Leiden Online Variation Database platform.
- Information on ARSA genotype could contribute to predict expected clinical variant and prognosis of pre-symptomatic patients, in the light of innovative therapeutic strategies.
Assessing the Pathogenicity of Insertion and Deletion Variants with the Variant Effect Scoring Tool (VEST-Indel)
- Pages: 28-35
- First Published: 07 October 2015
We further developed our Variant Effect Scoring Tool (VEST) to include classification of in-frame and frameshift indels (VEST-indel) as pathogenic or benign. VEST-indel includes a new feature for estimating the importance of a gene in human disease, and achieves improved specificity and balanced accuracy relative to existing methods. VEST is available as a standalone application, and as part of the CRAVAT webserver (cravat.us).
wKinMut-2: Identification and Interpretation of Pathogenic Variants in Human Protein Kinases
- Pages: 36-42
- First Published: 07 October 2015
wKinMut-2 is a tool for the identification and interpretation of variants affecting human protein kianases with a causative role in disease. The pathogenicity of variants is predicted using 9 different tools. Comprehensive information to understand the biological consequences of the variants is automatically mined, digested and homogenised. These two approaches combined facilitate the generation of working hypotheses to be confirmed with additional experiments.
Functional and Clinical Consequences of Novel α-Galactosidase A Mutations in Fabry Disease
- Pages: 43-51
- First Published: 29 September 2015

Mutations within the GLA gene cause Fabry disease (FD). Number of individuals with atypical disease outnumbers the classically affected (indicated by circle/oval size in the picture). The conventional classification “mutation vs. SNP” seems to be inappropriate in FD. We conclude that all, or at least the overall majority of gene variants can be risk factors for one or more FD symptoms.
Putative Prostate Cancer Risk SNP in an Androgen Receptor-Binding Site of the Melanophilin Gene Illustrates Enrichment of Risk SNPs in Androgen Receptor Target Sites
- Pages: 52-64
- First Published: 28 September 2015
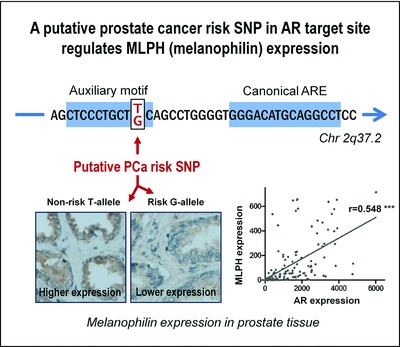
1490 primary androgen receptor (AR) target genes were identified in DuCaP prostate cancer cells. Prostate cancer risk SNPs and linked SNPs are significantly enriched in genomic AR target sites. A putative risk SNP in an AR binding site of the MLPH gene regulates melanophilin expression. More favorable tumor risk profile in prostate cancer patients with higher melanophilin expression suggest a tumor suppressive function of melanophilin.
A Role for Non-B DNA Forming Sequences in Mediating Microlesions Causing Human Inherited Disease
- Pages: 65-73
- First Published: 15 October 2015
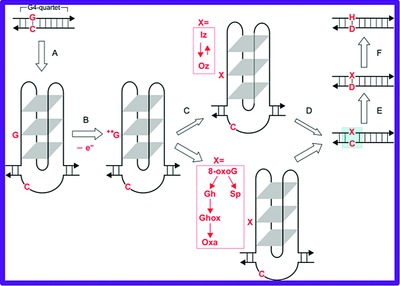
This is the first study assessing the extent to which four different types of non-B DNA-formingsequence direct, inverted, RY-rich mirror repeats, and G-quartets capable of slipped, hairpin/cruciform, triplex, and G-quadruplex structure formation, respectively, may be involved in mediating different types of micro-lesion causing (or associated with) human inherited disease. Novel mechanisms of mutagenesis, based on either the formation or resolution of non-B DNA structures, have been proposed providing support for a role for non-B DNA structures in mutagenesis.
Identification and Functional Characterization of CLCN1 Mutations Found in Nondystrophic Myotonia Patients
- Pages: 74-83
- First Published: 14 October 2015
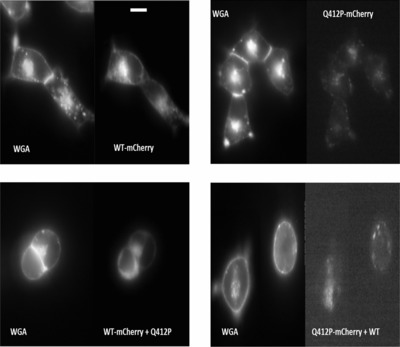
Clinical variability in myotonic conditions might lead to clinical misdiagnosis. Identification of the most likely disease causing-mutation will help to properly classify the probands diagnosed with any of these conditions. However, sometimes these mutations cannot fully explain atypical phenotypes, raising the possibility that other cell/molecular features of the disease are involved in that variability. Thus, this work was aimed to study a group of probands with atypical phenotypes and characterized electrophysiologically, the mutation found in the CLCN1 gene
Diagnostic Exome Sequencing Identifies a Novel Gene, EMILIN1, Associated with Autosomal-Dominant Hereditary Connective Tissue Disease
- Pages: 84-97
- First Published: 14 October 2015
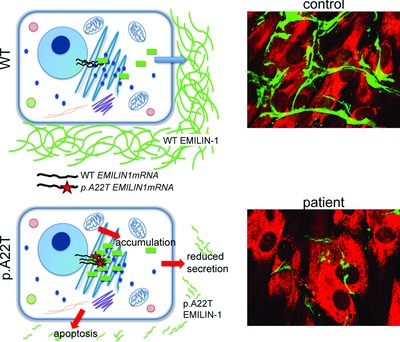
We identify a heterozygous genetic lesion in a family suffering from a connective tissue disorder resulting in the p.A22T missense mutation in the signal peptide cleavage site of EMILIN1. The mutation causes an impaired protein maturation leading to an abnormal accumulation in the endoplasmic reticulum, an increased apoptosis and a reduction in extracellular matrix deposition. This study proposes for the first time the association between a heritable EMILIN1 mutation and an autosomal-dominant connective tissue disease.
Alternative Splicing in the Human PMP22 Gene: Implications in CMT1A Neuropathy
- Pages: 98-109
- First Published: 21 October 2015
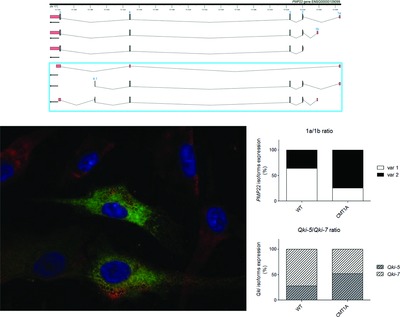
Genotype-phenotype correlations are not so obvious in PMP22-related CMT. We found three previously undescribed PMP22 human splicing variants specifically expressed in the PNS. Two of them encoded a new putative PMP22 protein isoform mainly expressed in the ER. Moreover, PMP22 alternative splicing is dysregulated in CMT1A and likely under the control of QKI. Overall our data suggest that an alteration of mRNA processing could be a pathogenic mechanism in CMT1A.
Efficient Generation of Gene-Modified Pigs Harboring Precise Orthologous Human Mutation via CRISPR/Cas9-Induced Homology-Directed Repair in Zygotes
- Pages: 110-118
- First Published: 07 October 2015
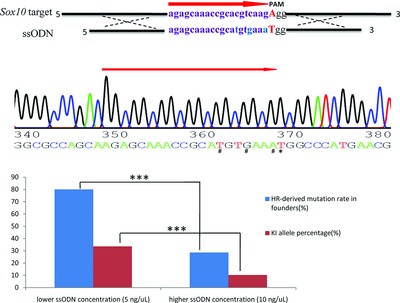
Without inhibition of non-homologous end joining (NHEJ) pathways and using single strand oligo DNA (ssODN) as templates, we efficiently generated gene-modified pigs harboring precise orthologous human mutation via CRISPR/Cas9-induced homology-directed repair (HDR) in zygotes with an efficiency as high as 80%. Besides, we found that a higher concentration of ssODN remarkably reduced HDR-derived mutation efficiency, suggesting that the activities of HDR relative to NHEJ were reduced with the higher amounts of ssODN.
Targeted Droplet-Digital PCR as a Tool for Novel Deletion Discovery at the DFNB1 Locus
- Pages: 119-126
- First Published: 07 October 2015
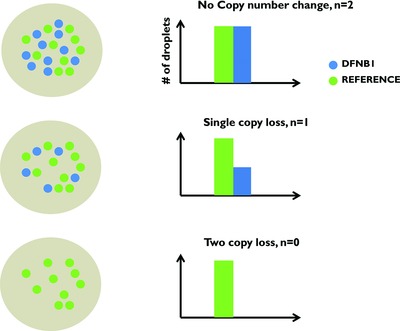
Unlike traditional PCR, targeted droplet-digital PCR is a robust, highly sensitive technology capable of detecting all reported, and potentially novel, DFNB1 copy number changes using a few probes. Here, we describe such an assay and the identification of a novel deletion at the DFNB1 locus.
A Standardized DNA Variant Scoring System for Pathogenicity Assessments in Mendelian Disorders
- Pages: 127-134
- First Published: 15 October 2015
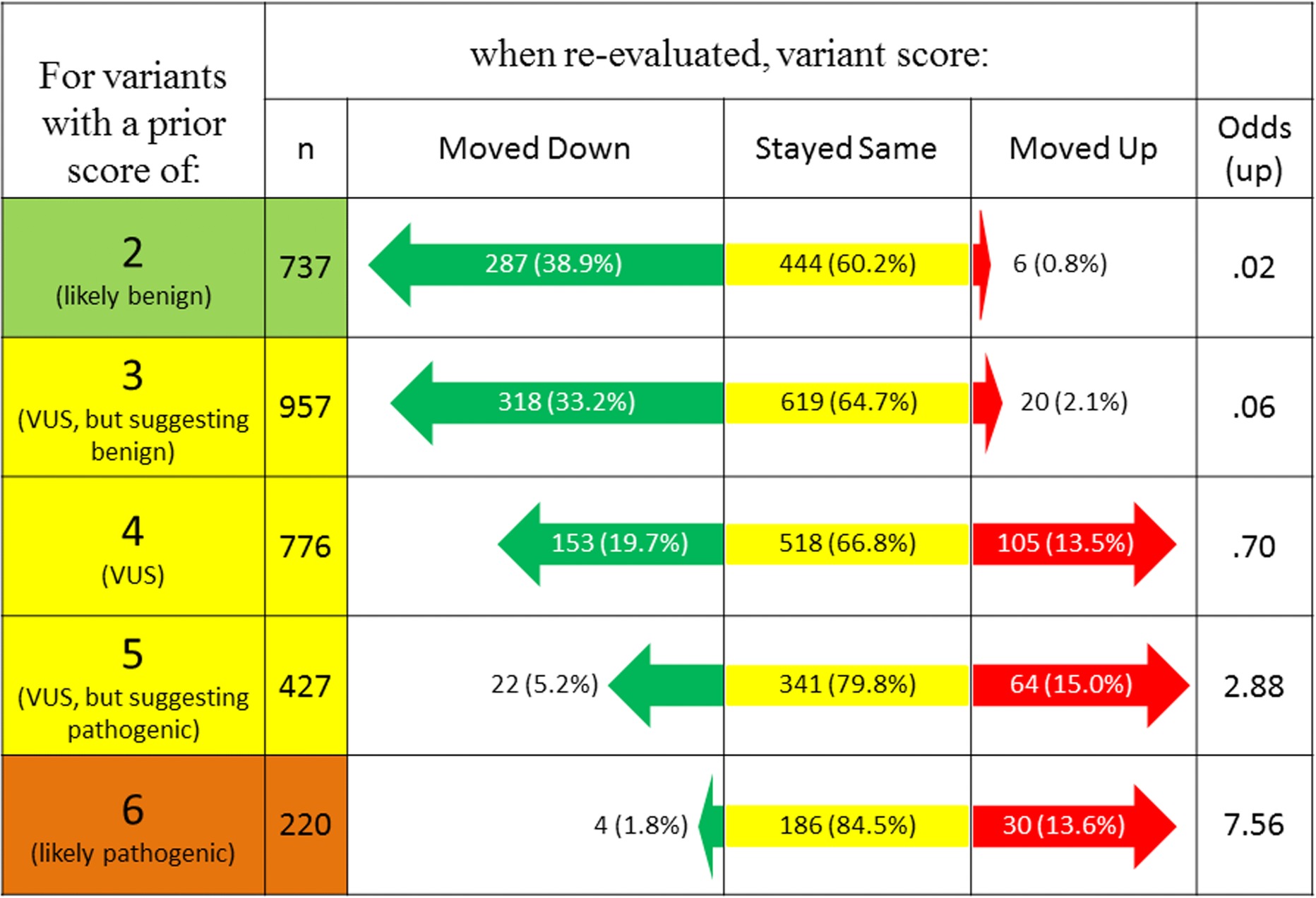
The article by Karbassi et al. describes a standardized DNA variant scoring system that uses multiple lines of evidence, three subclasses of VUS, technical training procedures, and weighted rules-based scoring to produce reliable and consistent clinical grade variant interpretations. This carefully curated scoring system provides the first roadmap to accurately process complex genetic information in diagnostics.