

Journal list menu

 Issue2013iv, 411-537
Issue2013iv, 411-537
Export Citations
Download PDFs
Human Variation 2.0: Using GWAS to Probe Intermediate Phenotypes
- Page: iv
- First Published: 05 March 2013
Pseudohypoparathyroidism Type Ia and Pseudo-Pseudohypoparathyroidism: The Growing Spectrum of GNAS Inactivating Mutations
- Pages: 411-416
- First Published: 05 March 2013
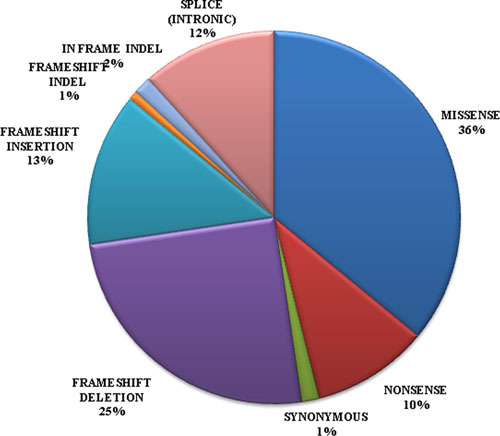
Pseudohypoprathyroidism (PHP) is a rare heterogeneous genetic disorder characterized by end-organ resistance to parathyroid hormone. Heterozygous inactivating GNAS mutations lead to either PHP type Ia or PPHP. The present work provides an updated collection of mutational and phenotypic data for both diagnostic and research purposes, as a step forward to a better understanding of PHP. The absence of genotype-phenotype correlation supports the hypothesis of Gsα haploinsufficiency as the molecular mechanism underlying PHP, independently of the mutation type/localization.
An Overview and Update of ATP7A Mutations Leading to Menkes Disease and Occipital Horn Syndrome
- Pages: 417-429
- First Published: 05 March 2013
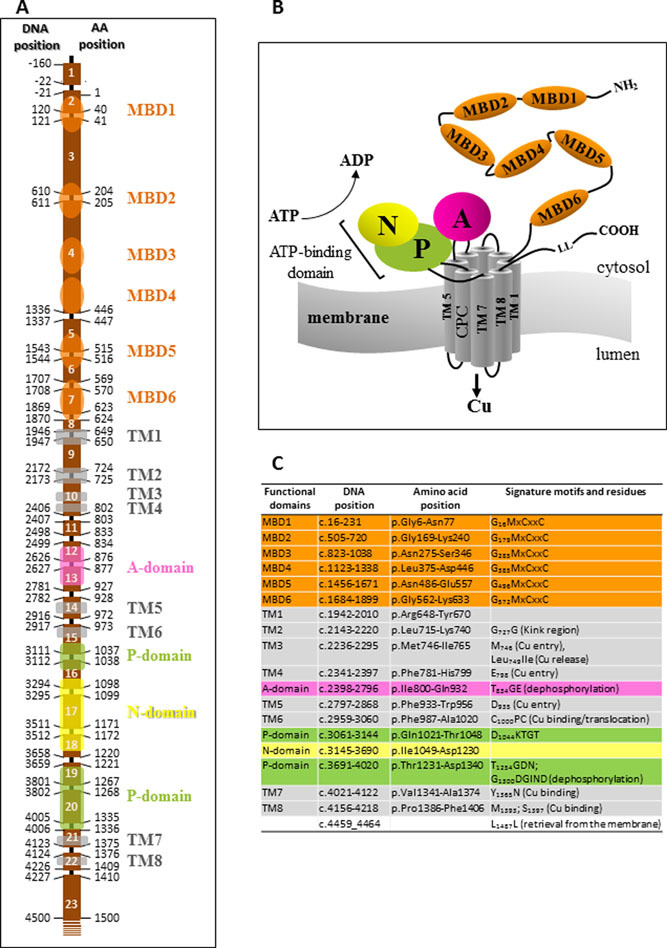
Menkes disease (MD) is an X-linked disorder of copper metabolism, characterized by progressive neurodegeneration and connective tissue abnormalities as well as typical steely hair. The defective gene, ATP7A, encodes a transmembrane protein, which is involved in the delivery of copper to the secreted cuproenzymes and in the export of surplus copper from cells. This study reviews 274 published and 18 novel ATP7A mutations identified in 370 unrelated MD patients, non-pathogenic variants, functional studies of the ATP7A mutations and animal models.
Exome Sequencing Identifies A Branch Point Variant in Aarskog–Scott Syndrome
- Pages: 430-434
- First Published: 05 March 2013
Identification of a Novel Oligomerization Disrupting Mutation in CRYΑA Associated with Congenital Cataract in a South Australian Family
- Pages: 435-438
- First Published: 05 March 2013

A p.R21Q mutation in the alpha-crystallin A gene was shown to segregate with a nuclear lamellar congenital cataract of variable severity in an Australian family. This mutation appears to affect the ability of the CRYAA protein to form the high molecular weight oligomers essential to its chaperone function.
Analysis of BRCA1 Variants in Double-Strand Break Repair by Homologous Recombination and Single-Strand Annealing
- Pages: 439-445
- First Published: 05 March 2013
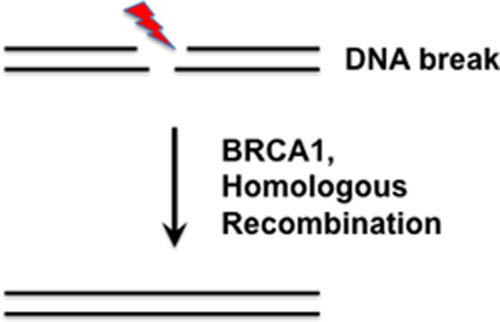
Missense substitutions of uncertain clinical significance in the BRCA1 gene are a vexing problem in genetic counseling for women who have a family history of breast or ovarian cancer. In this study, we evaluated whether functional assays for DNA repair can augment the genetic information. We found that the effects of BRCA1 missense substitutions on protein function in homologous recombination repair of DNA double strand breaks were predictive of disease association.
Mitochondrial Complex III Deficiency Caused by a Homozygous UQCRC2 Mutation Presenting with Neonatal-Onset Recurrent Metabolic Decompensation
- Pages: 446-452
- First Published: 05 March 2013
Whole exome sequencing combined with homozygosity mapping in a consanguineous family revealed that a homozygous missense mutation (p.Arg183Trp) in UQCRC2 causes a novel type of neonatal-onset recurrent metabolic decompensation (hypoglycemia, lactic acidosis, ketosis, and hyperammonemia). UQCRC2 encodes a core protein of complex III and the mutation results in instability of mitochondrial complex III and supercomplex with functional deficiency.
Drastic Effect of Germline TP53 Missense Mutations in Li–Fraumeni Patients
- Pages: 453-461
- First Published: 05 March 2013
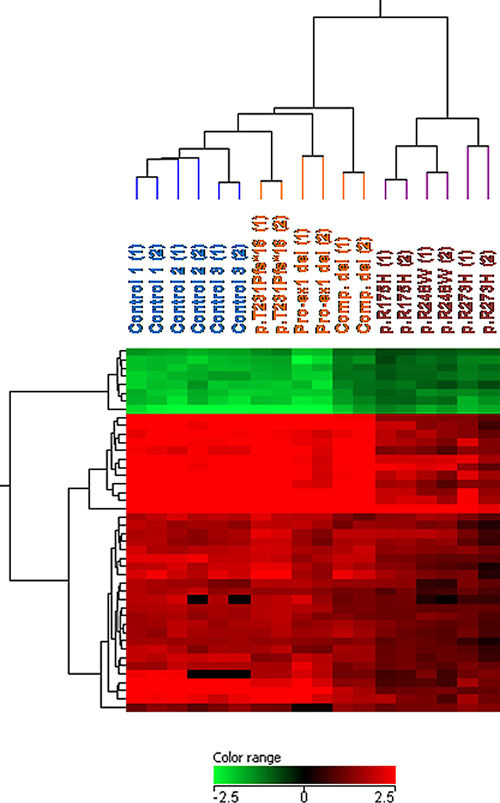
This manuscript describes a new functional assay of the p53 pathway, based on induction of DNA damage in EBV-immortalized lymphocytes, followed by comparative gene expression profiling. In lymphocytes from Li-Fraumeni (LFS) patients with canonical missense mutations, the number of induced genes and the level of known p53 target genes induction are strongly reduced, as compared with controls and LFS lymphocytes with null mutations. These results show that certain germline missense TP53 mutations, such as those with dominant negative effect, dramatically alter the response to DNA damage.
Mutations in CCDC39 and CCDC40 are the Major Cause of Primary Ciliary Dyskinesia with Axonemal Disorganization and Absent Inner Dynein Arms
- Pages: 462-472
- First Published: 05 March 2013
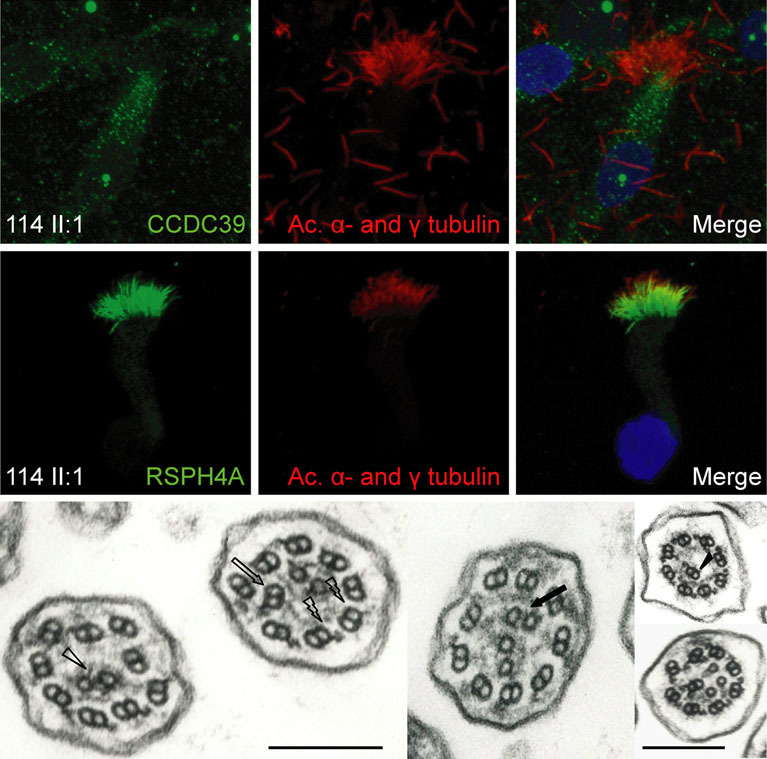
Around 12% of patients with primary ciliary dyskinesia (PCD), a recessively inherited ciliopathy caused by cilia/sperm dysmotility, have perturbed 9+2 microtubule cilia structure and inner dynein arm (IDA) loss. We find that biallelic CCDC39 and CCDC40 mutations cause 69% of this defect (37/54 families). We report 25 (19 novel) mutant alleles all causing predicted “null” alleles, with 73% homozygous mutations (27/37 families) including a major putative hotspot mutation, CCDC40 c.248delC. We propose the renaming this disorder to “IDA and microtubular disorganisation defect”.
Molecular Basis of Two-Exon Skipping (Exons 12 and 13) by c.1248+5g>a in OXCT1 Gene: Study on Intermediates of OXCT1 Transcripts in Fibroblasts
- Pages: 473-480
- First Published: 05 March 2013
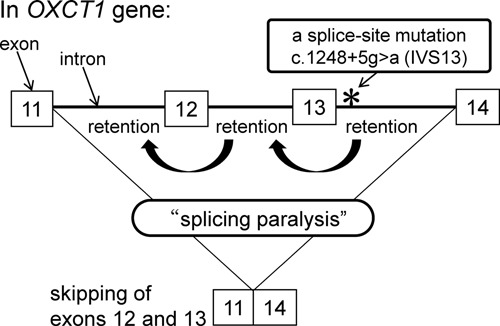
An IVS13 mutation (c.1248+5g>a) of the OXCT1 gene resulted predominantly in skipping of exons 12 and 13 in fibroblasts. Analysis of intermediates of OXCT1 transcripts in control fibroblasts showed that intron 11 was the last intron to be spliced and intron 12 removal occurred after intron 13 removal in the major pathway. Retention of the mutated intron 13 in the patient's fibroblasts causing retention of introns 12 and 11 resulted in the two exon skipping due to “splicing paralysis”.
Novel XPG (ERCC5) Mutations Affect DNA Repair and Cell Survival after Ultraviolet but not Oxidative Stress
- Pages: 481-489
- First Published: 05 March 2013
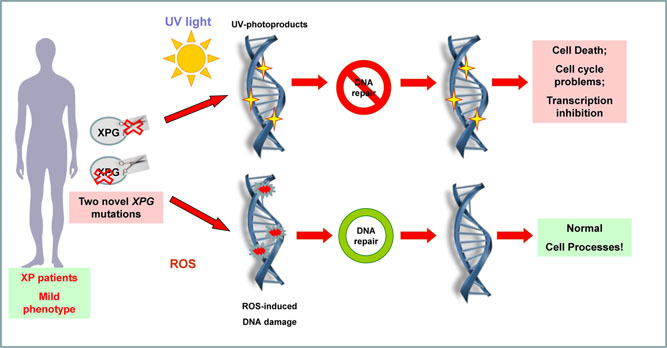
Two novel XPG mutations were detected from patients with mild xeroderma pigmentosum phenotype. Functional analyses demonstrate these novel mutations lead to important reduction capacity in the repair of UV-induced DNA lesions, but do not affect the repair of oxidatively generated DNA lesions.
Cancer Risks for MLH1 and MSH2 Mutation Carriers
- Pages: 490-497
- First Published: 05 March 2013
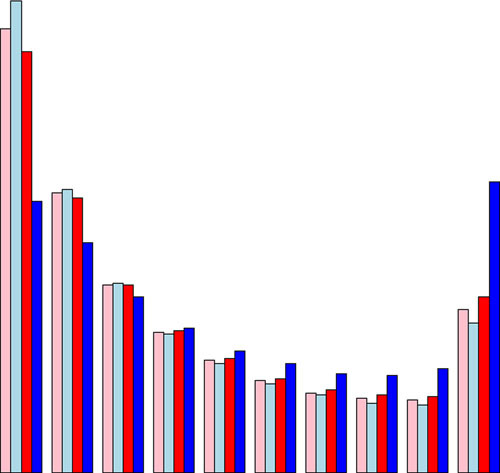
Our estimates of colorectal and endometrial cancer risks for MLH1 and MSH2 mutation carriers are the most precise currently available. Average risks are similar for the two genes but there is so much individual variation about the average that a sizeable proportion of carries are almost certain to develop cancer whereas another sizeable proportion only have population-level risks.
Delineation of the Clinical, Molecular and Cellular Aspects of Novel JAM3 Mutations Underlying the Autosomal Recessive Hemorrhagic Destruction of the Brain, Subependymal Calcification, and Congenital Cataracts
- Pages: 498-505
- First Published: 05 March 2013
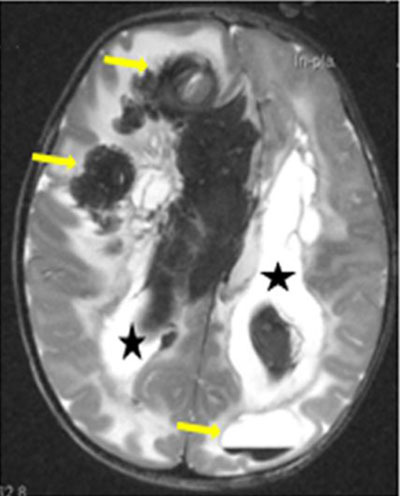
Mutations in Junctional adhesion molecule 3 (JAM3) cause the recessive syndrome involving hemorrhagic destruction of the brain, subependymal calcification, and congenital cataracts. T2-weighted axial brain MRI of a patient reveals multifocal intraparenchymal hemorrhages of varying ages leading to calcifications (arrows) as well as intraventricular hemorrhage (asterisks).
RP1L1 Variants are Associated with a Spectrum of Inherited Retinal Diseases Including Retinitis Pigmentosa and Occult Macular Dystrophy
- Pages: 506-514
- First Published: 05 March 2013
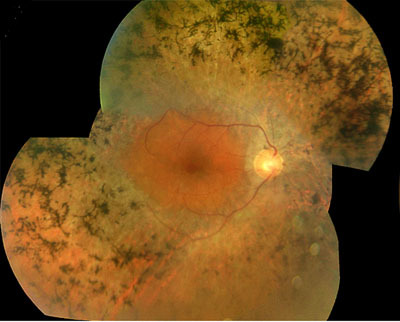
In this study we demonstrate that rare variants in RP1L1, encoding a photoreceptor specific protein, are associated with a range of inherited retinal disease. Our data suggest that presumed loss-of-function variants in the gene can cause autosomal recessive Retinitis Pigmentosa, whereas specific heterozygous missense variants are associated with a propensity to develop Occult Macular Dystrophy. The findings presented imply an important and diverse role for RP1L1 in human retinal physiology.
A Genome-Wide Assessment of Variability in Human Serum Metabolism
- Pages: 515-524
- First Published: 05 March 2013
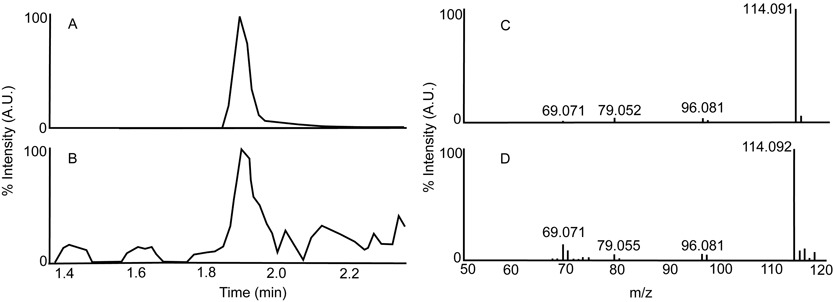
A genome-wide association study of metabolic quantitative traits (mQTLs) was conducted in Swedish men. A global non-targeted metabolite profiling approach was used resulting in the identification of both novel mQTLs and replication of previously described associations. Analyses involving biological pathways and a search for enrichment of mQTLs across the genome highlighted additional novel loci.
Comparative Analysis and Functional Mapping of SACS Mutations Reveal Novel Insights into Sacsin Repeated Architecture
- Pages: 525-537
- First Published: 05 March 2013

ARSACS is a neurological disease with mutations in SACS, encoding sacsin. Three repeated regions (Sacsin Internal RePeaTs) larger than the previously described Sacsin Repeating Region supra-domains were found in sacsin, organized in sub-repeats (sr1, sr2, sr3, srX). Comparative analysis of vertebrate sacsins in combination with fine positional mapping of a set of human mutations reveals that the sub-repeats are functional. The position of the pathogenic mutations in the sub-repeats appears to be related to the severity of the clinical phenotype.