

Bioassay-Guided Isolation and Structural Modification of the Anti-TB Resorcinols from Ardisia gigantifolia
Abstract
Tuberculosis (TB) is a highly contagious disease mainly caused by Mycobacterium tuberculosis H37RV. Antitubercular (anti-TB) bioassay-guided isolation of the CHCl3 extract of the leaves and stems of the medicinal plant Ardisia gigantifolia led to the isolation of two anti-TB 5-alkylresorcinols, 5-(8Z-heptadecenyl) resorcinol (1) and 5-(8Z-pentadecenyl) resorcinol (2). We further synthesized 15 derivatives based on these two natural products. These compounds (natural and synthetic) were evaluated for their anti-TB activity against Mycobacterium tuberculosis H37RV. Resorcinols 1 and 2 exhibited anti-TB activity with MIC values at 34.4 and 79.2 μm in MABA assay, respectively, and 91.7 and 168.3 μm in LORA assay, respectively. Among these derivatives, compound 8 was found to show improved anti-TB activity than its synthetic precursor (2) with MIC values at 42.0 μm in MABA assay and 100.2 μm in LORA assay. The active compounds should be regarded as new hits for further study as a novel class of anti-TB agents. The distinct structure–activity correlations of the parent compound were elucidated based on these derivatives.
Tuberculosis (TB) is a highly contagious bacterial disease most commonly manifesting as a pulmonary infection and mainly caused by Mycobacterium tuberculosis 1. The World Health Organization (WHO) estimates that there were about 11 million prevalent cases of TB in 2013, equivalent to 159 cases per 10 million population and lead up to 1.5 million deaths 2.
The drugs used for treating TB are more than 40 years old and are far from ideal. Drug-resistant TB (DR-TB) poses a major threat for the control of TB worldwide. In 2013, there were an estimated 480 000 new cases of multidrug-resistant TB (MDR-TB) worldwide and approximately 210 000 deaths 2. In the heavy MDR-TB burden countries, the average duration of hospital stay ranged from 7 to 240 days, with a median of 90 days 2. Two new drugs have been approved for the treatment of MDR-TB under specific conditions: bedaquiline and delamanid in 2012 3. However, these two drugs are the first compounds to be approved for use in TB treatment in nearly 40 years, and the only ones ever to be released specifically for the treatment of MDR-TB 4. This demands our continuous efforts to discover new anti-TB therapeutic agents that improve the treatment of multidrug-resistant and extensively drug-resistant strains and shortens the total duration of treatment.
Plant compounds, known for their enormous numbers and their remarkable structural diversity, are considered an excellent source for exploration of drug lead compounds and have received considerable attention as potential anti-TB agents 5, 6. Ardisia gigantifolia Stapf (Primulaceae; previously, Myrsinaceae) collected from Vietnam for the present research (see below) is a shrub growing in the shade and wet places of valley and hillsides and is widely distributed in Southeast Asia including Vietnam, Thailand, Malaysia, Indonesia, and Southern provinces of China 7, 8. The whole plant of this species has been used in folk medicine to eliminate blood stasis, disperse swelling, improve blood circulation, and also as an analgesic 9. This plant was investigated as part of our International Cooperative Biodiversity Group (ICBG) project, which was designed to address the related issues of biodiversity conservation, economic growth, and promotion of health through the discovery of anticancer, anti-HIV, antimalarial, and anti-TB natural products through collaboration with institutions in Vietnam, Laos, and the United States 10. This plant was found to be one of the first anti-TB plant leads in our efforts to discover anti-TB agents from plants of the tropical forests of Vietnam and Laos. The current article describes the isolation, structure elucidation, derivatization of the active natural products, and biological evaluation of the pharmacological activities of these compounds.
Experiment
General experimental procedures
NMR spectra were recorded on a Bruker DPX-300 MHz or a Bruker DPX-400 MHz spectrometer. Chemical shifts (δ) were expressed in ppm with reference to the solvent signals (CDCl3; 1H: 7.24 ppm; 13C: 77.00 pm), and coupling constants (J) were reported in Hz. All NMR experiments were obtained using standard pulse sequences supplied by the vendor. Column chromatography was carried out on silica gel (200–400 mesh, Natland International Corporation, Morrisville, NC, USA). Reversed-phase flash chromatography was accomplished with RP-18 silica gel (40–63 μm, EM Science). Thin-layer chromatography was performed on Whatman glass-backed plates coated with 0.25 mm layers of silica gel 60. HRTOF-MS spectra were recorded on a Micromass Q-TOF-2 spectrometer. All reagents were purchased from Sigma-Aldrich Chemical Co. (St Louis, MO, USA) and used without further purification. All solvents were reagent grade or HPLC grade.
Plant material
Leaf and stem sample (SVA0214) of A. gigantifolia was recollected in Cuc Phuong National Park, Nho Quan District, Ninh Binh Province, Vietnam, on October 21, 2001, from the same location where the original primary active sample (SV0214) was collected on March 20, 1999. The exact location was forest floor at northeast side of Bong at 500 m altitude, in a primary forest on a steep slope, 20o 21′ 13″ N, 105o 35′ 48″ E. It is a shrub 3 m tall, with the upper leaf surface dark green, lower surface greenish purple, the peduncle green, turning purple toward the tip, bearing purple flower buds with white top set on a purple pedicel. A voucher herbarium specimen of the recollected sample (Soejarto et al. 11809) and that of the primary sample (Soejarto et al. 10628) have been deposited at each of the following institutions: Cuc Phuong National Park Herbarium (CPNP) in Nho Quan, Ninh Binh, Vietnam; Herbarium of the Department of Botany (HN) of the Vietnam Academy of Science and Technology, Hanoi, Vietnam; and at the J. D. Searle Herbarium of the Field Museum (F), Chicago, USA.
Extraction and isolation
The dried and milled leaves and stems (5.2 kg) were extracted with CHCl3 (×3) to yield an extract (37.9 g), which was subsequently defatted with n-hexane and partitioned with CHCl3. The CHCl3-soluble fraction (33.0 g) was chromatographed over a silica gel column (400 g) and eluted by gradient elution with petroleum ether/EtOAc and EtOAc/MeOH to obtain eight fractions (F1-F8). Fraction F2 (5.15 g) demonstrated 91% inhibition against M. tuberculosis H37Rv at 50 μg/mL and was further chromatographed on a silica gel column (100 g) by gradient elution with CHCl3 and increasing concentration of Me2CO to yield six fractions (F9-F14). Fraction F11 demonstrated anti-TB activity against M. tuberculosis H37Rv with an MIC value of 12.5 μg/mL. This fraction (0.72 g) was subjected to flash column chromatography on a C-18 reverse phase (RP-18, 30 g) column. Subsequent gradient elution with H2O and increasing concentration of MeCN yielded 5-(8Z-heptadecenyl) resorcinol (1, 0.15 g) and 5-(8Z-pentadecenyl) resorcinol (2, 0.26 g) (Figure 1).

Preparation of the derivatives (3-14) of compounds 1 and 2
To a solution of compound 1 or 2 (5.0–8.0 mg) and corresponding selected acyl chloride or p-toluenesulfonyl chloride (TsCl) (3 eq) in CH2Cl2 (3 mL), triethylamine (TEA) (8 eq) and catalytic amount of 4-dimethylaminopyridine (DMAP) at 0 °C were added. The resulting reaction mixture was stirred at room temperature overnight. Volatile components in the reaction mixtures were removed by evaporation under reduced pressure, and the resulting residue was purified by silica gel column chromatography to afford ester derivatives 3-14, respectively.
Preparation of compounds 15-16 11
To a stirred suspension consisting of compound 14 (5.0 mg), silver acetate (AgOAc) (4.0 mg), and water (1.6 mg) in glacial acetic acid (5 mL), iodine (2.4 mg) was added. The resultant yellow mixture was stirred for 24 h at room temperature and then filtered through a cotton wool plug to remove insoluble material. The filtrate was poured into CH2Cl2 (20 mL) in a separatory funnel, which was washed successively with H2O (2 × 5 mL) and saturated aqueous sodium bicarbonate (NaHCO3) (5 mL). The aqueous layers were combined and extracted with CH2Cl2 (20 mL). The resulting organic layer was added to the original CH2Cl2 and further washed with brine (7 mL) and then dried with sodium sulfate (Na2SO4). Removal of the solvent under reduced pressure gave an orange residue.
A solution of the orange residue, potassium carbonate (K2CO3, 5.5 mg) in MeOH, and H2O (5 mL, VMeOH: 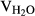 = 10:1) was stirred at room temperature overnight. The solvent was removed under reduced pressure, and the residue was subjected to silica gel column chromatography to give diols 15-16.
= 10:1) was stirred at room temperature overnight. The solvent was removed under reduced pressure, and the residue was subjected to silica gel column chromatography to give diols 15-16.
Preparation of compound 17 12
Sodium periodate (NaIO4, 1.29 g, 6.0 mmol) was dissolved in 2.5 mL of hot water (~70 °C). To this hot solution, silica gel (230–400 mesh, 5 g) was added with vigorous swirling and shaking to afford a free-flowing powder.
To a vigorously stirred suspension of this silica gel-supported NaIO4 reagent (9 mg) in CH2Cl2 (2 mL), a solution of diol 16 (3 mg) in CH2Cl2 (3 mL) was added. The mixture was stirred for 30 min and then filtered through a sintered glass funnel. The retained silica gel was thoroughly washed with CH2Cl2 (3 × 10 mL) and added to the filtrate. In vacuo, removal of the organic solvent from the filtrate afforded aldehyde 17.
Compound 3, Amount, 2.5 mg; yield, 85%; colorless gum; 1H NMR (Figure S1) (400 MHz, CDCl3) δ: 8.04 (4H, d, J = 9.1 Hz), 6.93 (3H, brs), 6.69 (4H, d, J = 9.1 Hz), 5.38–5.30 (2H, m), 3.08 (12H, s), 2.64 (2H, t, J = 7.6 Hz), 2.05–1.97 (4H, m), 1.68–1.59 (2H, m), 1.38–1.22 (20H, m), 0.88 (3H, t, J = 7.0 Hz); 13C NMR (Figure S2) (100 MHz, CDCl3) δ: 165.3, 153.6, 151.5, 145.1, 132.0, 131.2, 129.9, 129.8, 118.9, 115.9, 113.2, 110.7, 110.6, 40.0, 35.8, 31.9, 31.8, 31.0, 29.7, 29.5, 29.4, 29.3, 29.0, 27.2, 22.6, 14.1; HRTOF-positive ESIMS m/s 641.4316 [M + 1]+ (calcd for C41H57N2O4, 641.4313).
Compound 4, Amount, 3.0 mg; yield, 90%; colorless gum; 1H NMR (Figure S3) (400 MHz, CDCl3) δ: 8.04 (4H, d, J = 8.9 Hz),6.93 (3H, brs), 6.69 (4H, d, J = 9.0 Hz), 5.38–5.32 (2H, m), 3.08 (12H, s), 2.64 (2H, t, J = 7.9 Hz), 2.05–1.97 (4H, m), 1.68–1.61 (2H, m), 1.33–1.28 (16H, m), 0.88 (3H, t, J = 7.0 Hz); 13C NMR (Figure S4) (100 MHz, CDCl3) δ: 165.3, 153.7, 151.6, 145.0, 132.0, 129.9, 118.9, 115.9, 113.3, 110.7, 40.0, 35.8, 31.8, 31.0, 29.8, 29.7, 29.4, 29.3, 29.2, 29.0, 27.2, 22.7, 14.1; HRTOF-positive ESIMS m/s 613.3973 [M + 1]+ (calcd for C39H53N2O4, 613.4000).
Compound 5, Amount, 2.0 mg; yield, 70%; colorless gum; 1H NMR (Figure S5) (400 MHz, CDCl3) δ: 7.15 (2H, dd, J = 4.0, 1.8 Hz), 6.91 (3H, brs), 6.88 (2H, dd, J = 2.2, 2.2 Hz), 6.19 (2H, dd, J = 4.1, 2.5 Hz), 5.36–5.32 (2H, m), 3.96 (6H, s), 2.63 (2H, t, J = 7.6 Hz), 2.08–1.93 (4H, m), 1.69–1.58 (2H, m), 1.38–1.22 (20H, m), 0.88 (3H, t, J = 6.2 Hz); 13C NMR (Figure S6) (100 MHz, CDCl3) δ: 159.3, 150.8, 145.3, 130.6, 129.9, 121.4, 119.2, 119.0, 113.3, 108.3, 36.9, 35.7, 31.7, 31.0, 29.7, 29.6, 29.5, 29.4, 29.3, 29.0, 27.2, 22.6, 14.1; HRTOF-positive ESIMS m/s 561.3660 [M + 1]+ (calcd for C35H49N2O4, 561.3687).
Compound 6, Amount, 1.8 mg; yield, 75%; colorless gum; 1H NMR (Figure S7) (400 MHz, CDCl3) δ: 7.16 (2H, dd, J = 4.0, 1.8 Hz), 6.91 (3H, brs), 6.88 (2H, dd, J = 2.2, 2.2 Hz), 6.19 (2H, dd, J = 4.0, 2.5 Hz), 5.35 (2H, m), 3.96 (6H, s), 2.63 (2H, t, J = 7.5 Hz), 2.08–1.96 (4H, m), 1.69–1.59 (2H, m), 1.41–1.10 (16H, m), 0.88 (3H, t, J = 7.0 Hz); 13C NMR (Figure S8) (100 MHz, CDCl3) δ: 159.3, 150.8, 145.2, 130.6, 129.9, 129.8, 121.4, 119.2, 119.0, 113.3, 108.3, 36.9, 35.7, 31.7, 30.9, 29.7, 29.3, 29.2, 29.0, 27.2, 22.6, 14.1; HRTOF-positive ESIMS m/s 533.3376 [M + 1]+ (calcd for C33H45N2O4, 533.3374).
Compound 7, Amount, 1.5 mg; yield, 65%; colorless gum; 1H-NMR (Figure S9) (400 MHz, CDCl3) δ: 9.40 (2H, d, J = 1.5 Hz), 8.87 (2H, dd, J = 4.8, 1.5 Hz), 8.51–8.40 (2H, m), 7.48 (2H, dd, J = 8.0, 4.8 Hz), 7.05 (1H, d, J = 1.9 Hz), 7.03 (2H, brs), 5.38–5.30 (2H, m), 2.69 (2H, t, J = 7.8 Hz), 2.04–1.98 (4H, m), 1.71–1.62 (2H, m), 1.40–1.21 (20H, m), 0.88 (3H, t, J = 7.1 Hz); 13C NMR (Figure S10) (100 MHz, CDCl3) δ: 163.5, 154.1, 151.4, 150.7, 146.0, 137.6, 129.9, 129.8, 125.4, 123.5, 119.3, 112.7, 35.8, 31.7, 30.9, 29.7, 29.6, 29.5, 29.4, 29.3, 29.2, 29.0, 27.2, 22.6, 14.1; HRTOF-positive ESIMS m/s 557.3367 [M + 1]+ (calcd for C35H45N2O4, 557.3374).
Compound 8, Amount, 2.0 mg; yield, 60%; colorless gum; 1H NMR (Figure S11) (400 MHz, CDCl3) δ: 9.39 (2H, s), 8.87 (2H, d, J = 4.8 Hz), 8.46 (2H, d, J = 8.0 Hz), 7.49 (2H, dd, J = 7.8, 4.9 Hz), 7.05 (1H, brs), 7.03 (2H, brs), 5.47–5.20 (2H, m), 2.69 (2H, t, J = 7.8 Hz), 2.10–1.94 (4H, m), 1.74–1.56 (2H, m), 1.42–1.18 (16H, m), 0.87 (3H, t, J = 6.3 Hz); 13C NMR (Figure S12) (100 MHz, CDCl3) δ: 163.5, 154.1, 151.3, 150.7, 146.0, 137.6, 130.0, 129.7, 125.4, 123.5, 119.3, 112.7, 35.7, 31.7, 30.9, 29.7, 29.3, 29.2, 29.1, 28.9, 27.2, 27.1, 22.6, 14.1; HRTOF-positive ESIMS m/s 529.3061 [M + 1]+ (calcd for C33H41N2O4, 529.3062).
Compound 9, Amount, 2.4 mg; yield, 85%; colorless gum; 1H NMR (Figure S13) (400 MHz, CDCl3) δ: 8.88 (4H, d, J = 5.1 Hz), 8.01 (4H, d, J = 5.9 Hz), 7.03 (1H, brs), 7.02 (2H, brs), 5.42–5.26 (2H, m), 2.68 (2H, t, J = 7.8 Hz), 2.12–1.91 (4H, m), 1.71–1.58 (2H, m), 1.37–1.23 (20H, m), 0.88 (3H, t, J = 6.5 Hz); 13C NMR (Figure S14) (100 MHz, CDCl3) δ: 163.4, 150.9, 150.7, 146.2, 136.6, 129.9, 129.8, 123.2, 119.3, 112.5, 35.8, 31.8, 30.9, 29.7, 29.5, 29.4, 29.3, 29.2, 29.0, 27.7, 27.2, 22.6, 14.1; HRTOF-positive ESIMS m/s 557.3371 [M + 1]+ (calcd for C35H45N2O4, 557.3374).
Compound 10, Amount, 1.2 mg; yield, 30%; colorless gum; 1H-NMR (Figure S15) (400 MHz, CDCl3) δ: 8.86 (2H, d, J = 5.7 Hz), 8.00 (2 H, dd, J = 4.4, 1.6 Hz), 6.62 (1H, dd, J = 1.8, 1.8 Hz), 6.61 (1H, dd, J = 2.0, 2.0 Hz), 6.56 (1 H, dd, J = 2.2, 2.2 Hz), 5.38–5.32 (2H, m), 5.22 (1 H, brs), 2.58 (2H, t, J = 7.6 Hz), 2.04–1.98 (4H, m), 1.66–1.59 (2H, m), 1.36–1.23 (16H, m), 0.88 (3H, t, J = 6.9 Hz); HRTOF-positive ESIMS m/s 424.2836 [M + 1]+ (calcd for C27H38NO3, 424.2846).
Compound 11, Amount, 3.0 mg; yield, 95%; colorless gum; 1H-NMR (Figure S16) (400 MHz, CDCl3) δ: 8.20 (4H, d, J = 7.6 Hz), 7.64 (2H, dd, J = 7.4, 7.4 Hz), 7.52 (4H, dd, J = 7.7, 7.7 Hz), 7.00 (1H, d, J = 1.7 Hz), 6.99 (2H, brs), 5.45–5.26 (2H, m), 2.67 (2H, t, J = 7.9 Hz), 2.06–1.96 (4H, m), 1.69–1.62 (2H, m), 1.37–1.23 (20H, m), 0.88 (3H, t, J = 6.5 Hz); 13C-NMR (Figure S17) (100 MHz, CDCl3) δ: 164.9, 151.2, 145.6, 133.6, 130.2, 129.9, 129.4, 128.6, 119.1, 112.9, 35.8, 31.7, 31.0, 29.7, 29.5, 29.4, 29.3, 29.0, 27.2, 22.6, 14.1; HRTOF-positive ESIMS m/s 577.3301 [M + Na]+ (calcd for C37H46NaO4, 577.3288).
Compound 12, Amount, 8 mg; yield, 90%; colorless gum; 1H-NMR (Figure S18) (400 MHz, CDCl3) δ: 8.20 (4H, dd, J = 7.1, 1.4 Hz), 7.67–7.60 (2H, m), 7.51 (4H, dd, J = 7.4, 7.4 Hz), 7.00 (1H, d, J = 2.0 Hz), 6.99 (2H, d, J = 2.0 Hz), 5.40–5.30 (2H, m), 2.69 (2H, t, J = 7.7 Hz), 2.07–1.95 (4H, m), 1.70–1.61 (2H, m), 1.41–1.21 (16H, m), 0.87 (3H, t, J = 6.9 Hz); HRTOF-positive ESIMS m/s 527.3145 [M + 1]+ (calcd for C35H43O4, 527.3156).
Compound 13, Amount, 2.2 mg; yield, 85%; colorless gum; 1H NMR (Figure S19) (400 MHz, CDCl3) δ: 7.64 (4H, d, J = 8.2 Hz), 7.31 (4H, d, J = 8.1 Hz), 6.69 (2H, brs), 6.45 (1H, brs), 5.46–5.28 (2H, m), 2.69–2.40 (8H, m), 2.10–1.94 (4H, m), 1.40–1.20 (22H, m), 0.88 (3H, t, J = 6.1 Hz); 13C NMR (Figure S20) (100 MHz, CDCl3) δ: 149.4, 146.0, 145.6, 131.9, 129.9, 129.8, 128.4, 121.1, 114.1, 35.3, 31.7, 30.7, 29.7, 29.5, 29.4, 29.3, 29.0, 27.2, 22.6, 21.7, 14.1; HRTOF-positive ESIMS m/s 677.2931 [M + Na]+ (calcd for C37H50NaO6S2, 677.2941).
Compound 14, Amount, 7.5 mg; yield, 90%; colorless gum; 1H NMR (Figure S21) (400 MHz, CDCl3) δ: 7.64 (4H, d, J = 8.3 Hz), 7.32 (4H, d, J = 8.0 Hz), 6.70 (2H, d, J = 2.2 Hz), 6.45 (1H, dd, J = 2.2, 2.2 Hz), 5.41–5.30 (2H, m), 2.46–2.42 (8H, m), 2.10–1.95 (4H, m), 1.43–1.10 (18H, m), 0.88 (3H, t, J = 7.0 Hz); HRTOF-positive ESIMS m/s 627.2800 [M + H]+ (calcd for C35H47O6S2, 627.2809).
Compound 15, Amount, 1.1 mg; yield, 25%; colorless gum; 1H-NMR (Figure S22) (400 MHz, CDCl3) δ: 7.72 (2H, d, J = 8.3 Hz), 7.31 (2H, d, J = 8.0 Hz), 6.54 (1H, d, J = 1.8 Hz), 6.34 (2H, d, J = 1.7 Hz), 5.61 (1H, brs), 3.67–3.56 (2H, m), 2.50–2.39 (5H, m), 2.01–1.85 (2H, m), 1.52–1.41 (4H, m), 1.36–1.19 (16H, m), 0.88 (3H, t, J = 6.8 Hz); HRTOF-positive ESIMS m/s 529.2585 [M + Na]+ (calcd for C28H42O6NaS, 529.2594).
Compound 16, Amount, 3.5 mg; yield, 35%; colorless gum; 1H-NMR (Figure S23) (400 MHz, CDCl3) δ: 7.65 (4H, d, J = 8.3 Hz), 7.32 (4H, d, J = 8.1 Hz), 6.72 (2H, d, J = 2.1 Hz), 6.44 (1H, dd, J = 2.2, 2.2 Hz), 3.64–3.57 (2H, m), 2.48–2.41 (8H, m), 1.87–1.78 (2H, m), 1.46–1.38 (4H, m), 1.33–1.18 (16H, m), 0.88 (3H, t, J = 6.7 Hz); HRTOF-positive ESIMS m/s 683.2679 [M + Na]+ (calcd for C35H48NaO8S2, 683.2683).
Compound 17, Amount, 1.5 mg; yield, 95%; colorless gum; 1H-NMR (Figure S24) (400 MHz, CDCl3), δ: 9.77 (1H, t, J = 1.8 Hz), 7.65 (4H, d, J = 8.4 Hz), 7.30 (4H, d, J = 8.0 Hz), 6.72 (2H, d, J = 2.2 Hz), 6.43 (1H, dd, J = 2.2, 2.2 Hz), 2.46–2.41 (10H, m), 1.64–1.60 (2H, m), 1.42–1.36 (2H, m), 1.31–1.23 (6H, m); 13C NMR (Figure S25) (100 MHz, CDCl3) δ: 202.8, 149.5, 145.8, 145.7, 132.0, 129.9, 128.5, 121.2, 114.1, 43.9, 35.3, 30.6, 29.1, 29.0, 28.7, 22.0, 21.8; HRTOF-positive ESIMS m/s 545.1657 [M + H]+ (calcd for C28H32O7S2, 545.1662).
Anti-TB activity bioassays
Extracts, fractions, purified compounds, and derivatives were subjected to in vitro assays. Primary screening was conducted at 100 μg/mL against M. tuberculosis H37Rv (ATCC 27294) using the microplate Alamar blue assay (MABA) and low-oxygen recovery assay (LORA), according to the procedures described by Collins 13 and Cho 14, respectively. Samples showing ≥90% inhibition in the primary screening were considered active and then retested at lower concentrations against M. tuberculosis H37Rv to determine the actual MIC. The MIC is defined as the lowest concentration effecting a reduction in fluorescence or luminescence of 90% with respect to untreated controls.
Results and discussion
Isolation of resorcinols 1 and 2
The CHCl3 extract made from the initially collected leaves and stems of A. gigantifolia demonstrated anti-TB activity with an MIC value of 25 μg/mL. A larger quantity of the leaf and stem samples was subsequently recollected from the same location to isolate the active compounds. The dried sample (5.2 kg) was milled and extracted with CHCl3, followed by in vacuo evaporation to afford a dried extract (37.9 g). Through bioassay-guided fractionation of the CHCl3 extract by repeated column chromatography on silica gel, fraction F11 was identified as the anti-TB fraction, with an MIC value of 12.5 μg/mL against M. tuberculosis H37Rv. Further separation of F11 using RP-18 silica gel led to the isolation of the anti-TB compounds 5-(8Z-heptadecenyl) resorcinol (1) and 5-(8Z-pentadecenyl) resorcinol (2) (Figure 1).
Compounds 1 and 2 were obtained as colorless gums and showed very similar NMR data (Table 1), suggesting that they have similar structures. Both compounds contain an aromatic ring, a C-C double bond, multimethylenes, and a methyl as evidenced by the 1H and 13C-NMR spectral data. Compound 1 was shown to have 14 methylenes, two methylenes more than 2 according to the analysis of the HRTOF-MS (1: [M-H]− m/z 345.2789, calcd. 345.2799, C23H37O2; 2: [M-H]− m/z 317.2483, calcd. 317.2486, C21H33O2) and the NMR data. The coupling patterns in the downfield region [1: δH 6.18 (1H, brs, 4-H), 6.24 (2H, brs, 2, 6-H); 2: δH 6.17 (1H, brs, 4-H), 6.25 (2H, brs, 2, 6-H)] showed that both compounds have a 1, 3, 5-substituted benzene ring. Compounds 1 and 2 were determined to be a 5-alkylresorcinols with a double bond in the side chain, based on the above data. In comparison with the literature data, 1 and 2 were thus identified as 5-(8Z-heptadecenyl) resorcinol and 5-(8Z-pentadecenyl) resorcinol, respectively 11, 15-17.
| Position | 1 | 2 | ||
|---|---|---|---|---|
| δH (mult)a | δC (mult)b | δH (mult)a | δC (mult)b | |
| 1 | 156.9 s | 156.4 s | ||
| 2 | 6.18 (1H, brs) | 100.2 d | 6.17 (1H, brs) | 100.2 d |
| 3 | 156.9 s | 156.4 s | ||
| 4 | 6.24 (2H, brs) | 108.1 d | 6.25 (2H, brs) | 108.1 d |
| 5 | 146.2 s | 146.2 s | ||
| 6 | 6.24 (2H, brs) | 108.1 d | 6.25 (2H, brs) | 108.1 d |
| 1′ | 2.35 (2H, t, 7.5) | 35.84 t | 2.45 (2H, t, 7.5) | 35.84 t |
| 2′ | 1.56 (2H, brs) | 31.79 t | 1.54 (2H, brs) | 31.79 t |
| 3′ | 1.27 (10 × 2H, m) | 29.31 t | 1.29 (8 × 2H, m) | 29.29 t |
| 4′ | 1.27 (10 × 2H, m) | 29.77 t | 1.29 (8 × 2H, m) | 29.41 t |
| 5′ | 1.27 (10 × 2H, m) | 29.77 t | 1.29 (8 × 2H, m) | 29.74 t |
| 6′ | 1.27 (10 × 2H, m) | 31.06 t | 1.29 (8 × 2H, m) | 29.74 t |
| 7′ | 2.01 (2 × 2H, m) | 27.23 t | 1.29 (8 × 2H, m) | 31.05 t |
| 8′ | 5.35 (2 × 1H, m) | 129.9 d | 2.01 (2 × 2H, m) | 27.24 t |
| 9′ | 5.35 (2 × 1H, m) | 129.9 d | 5.34 (2 × 1H, m) | 130.0 t |
| 10′ | 2.01 (2 × 2H, m) | 27.23 t | 5.34 (2 × 1H, m) | 129.8 t |
| 11′ | 1.27 (10 × 2H, m) | 31.06 t | 2.01 (2 × 2H, m) | 27.24 t |
| 12′ | 1.27 (10 × 2H, m) | 29.77 t | 1.29 (8 × 2H, m) | 29.41 t |
| 13′ | 1.27 (10 × 2H, m) | 29.77 5 | 1.29 (8 × 2H, m) | 31.94 t |
| 14′ | 1.27 (10 × 2H, m) | 29.56 t | 1.29 (8 × 2H, m) | 22.67 t |
| 15′ | 1.27 (10 × 2H, m) | 31.94 t | 0.88 (3H, t, 6.2) | 14.5 q |
| 16′ | 1.27 (10 × 2H, m) | 22.67 t | ||
| 17′ | 0.88 (3H, t, 6.2) | 14.5 q | ||
- a Multiplicities in parentheses represent: brs (broad singlet), d (doublet), m (multiplicity), t (triplet).
- b Multiplicities represent: s (quaternary carbon), d (CH), t (CH2), and q (CH3).
Preparation of resorcinols 1 and 2 derivatives
In an attempt to improve the biological activity of the isolated natural resorcinols, we initiated a structural modification effort. To that end, 15 derivatives were prepared by esterification of the phenolic hydroxyl groups and hydroxylation of the double bonds of compounds 1 and 2. As depicted in Scheme 1, the phenolic hydroxyl groups of the resorcinols were esterified with aromatic acyl chlorides including heterocyclic carbonyl chlorides to afford ester derivatives 3-14 in 30–95% yield. The diester 9 was prepared by treatment of 1 with 3 equivalents of isonicotinic acid chloride, but the monoester 10 was obtained by treatment of the resorcinol with 1.2 equivalents of isonicotinic acid chloride in a yield of 30%.

As shown in Scheme 2, compound 14 was subjected to a Woodward–Prevost reaction [14], followed by the subsequent hydrolysis using K2CO3, to yield derivatives 15-16. Compound 15 was obtained due to the deprotection of the intramolecular hydroxyl groups in the presence of K2CO3. Further oxidative cleavage of the diol group of 16 with NaIO4 gave aldehyde 17.

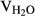 = 20:1; (b) K2CO3, CH3OH/H2O; (c) NaIO4·SiO2, CH2Cl2.
= 20:1; (b) K2CO3, CH3OH/H2O; (c) NaIO4·SiO2, CH2Cl2.Anti-TB activity
The source plant extract was identified as an anti-TB lead through our screening effort, and compounds 1-2 were subsequently isolated through bioassay-directed separation by determining MICs against replicating and non-replicating M. tuberculosis H37Rv using the MABA and LORA, respectively. Compound 1 showed MIC values of 34.4 μm against replicating cultures and 91.7 μm against non-replicating cultures, and 2 had MIC values of 79.2 μm against replicating cultures and 168.3 μm in against non-replicating cultures (Table 2).
| Compound | Inhibition MABA at 100 μg/mL | Inhibition LORA at 100 μg/mL | MIC MABA μg/mL (μm) | MIC LORA μg/mL (μm) |
|---|---|---|---|---|
| 1 | – | – | 11.9 (34.4) | 31.7 (91.7) |
| 2 | – | – | 25.2 (79.2) | 53.5 (168.3) |
| 3 | 0% | 0% | >100 | >100 |
| 4 | 4% | 0% | >100 | >100 |
| 5 | 0% | 6% | >100 | >100 |
| 6 | 15% | 13% | >100 | >100 |
| 7 | 88% | 64% | >100 | >100 |
| 8 | – | – | 22.2 (42.0) | 52.9 (100.2) |
| 9 | 87% | 61% | >100 | >100 |
| 10 | – | – | 42.3 (100.0) | 91.4 (216.2) |
| 11 | 0% | 9% | >100 | >100 |
| 12 | 10% | 36% | >100 | >100 |
| 13 | 16% | 38% | >100 | >100 |
| 14 | 26% | 46% | >100 | >100 |
| 15 | 89% | – | >100 | 90.5 (178.9) |
| 16 | 42% | 49% | >100 | >100 |
| 17 | 18% | 11% | >100 | >100 |
| Rifampin | (0.06) | (0.24) | ||
| Isoniazid | (0.47) | (>256) | ||
| Metronidazole | (>512) | (31.2) | ||
| Capreomycin | (3.51) | (3.73) | ||
| Streptomycin | (0.57) | (0.88) |
- a Minimum inhibitory concentration (MIC), determined under aerobic (MABA) or hypoxic (LORA) conditions against Mycobacterium tuberculosis H37Rv. Each value is the mean of at least three independent determinations.
In addition to the natural occurring resorcinols (1 and 2), we prepared 15 synthetic derivatives of these molecules for assessment of anti-TB potentials. The synthetic compounds were evaluated for their anti-TB activities against M. tuberculosis H37Rv in vitro (Table 2). While most of the derivatives displayed little or no inhibitory activity against the bacteria at the concentration of 100 μg/mL, derivative 8 showed equivalent activity to that of compound 1 with the MIC values at 42.0 μm in MABA assay and 100.2 μm in LORA assay. Through analysis of the activity data of Table 2, distinct structure–activity relationships (SARs) have been observed for these resorcinol compounds. Based on the SAR analysis, we obtained some preliminary conclusion: (i) Although the esterification approach did not significantly boost the activity, the slight improvement of the anti-TB activity of 8 in comparison with its parent compound (2) indicated that the phenolic hydroxyl groups may be used as the functional groups to synthesize other derivatives; (ii) Presence of the double bond in the side chain is essential to retain the anti-TB activity for this type of compounds. This effect was observed when the double bond was hydrolyzed as in the cases of compounds 15-16; (iii) the C2 symmetrical structure may not be important for the anti-TB activity as evidenced by the cases of compounds 9 and 10.
Although the 15 resorcinol derivatives synthesized did not produce a significant improvement in the anti-TB activity of compounds 1 and 2, the activity profiles of compound 8 verified that the anti-TB activity was marginally enhanced by our present synthetic approach. Further, as 8 contains nitrogen, it can be made water soluble by preparing it as a salt compound, which is worthy for further study as a novel anti-TB agent.
There have been only two anti-TB drugs introduced in the past 40 years, and the rapid acquisition of drug resistance to the existing drugs necessitates development of new, effective, and affordable anti-TB drugs 4. Plant-derived anti-TB compounds provide a great potential for discovery of novel anti-TB agents due to their exceptionally wide diversified structure classes, including terpenoids, alkaloids, phenolic compounds, and so on 18. Our bioassay-guided fractionation of the leaves and stems of the medicinal plant A. gigantifolia led to the isolation of two active resorcinols (1 and 2), which demonstrated inhibitory activity against M. tuberculosis (H37RV) in vitro with MIC values at 34.4 and 79.2 μm in MABA assay, respectively, and 91.6 and 168.2 μm in LORA assay, respectively. Hence, medicinal plants remain an important resource to find new therapeutic agents.
Conclusions
In conclusion, anti-TB bioassay-guided fractionation of the CHCl3 extract of the leaves and stems of A. gigantifolia led to the isolation of two 5-alkylresorcinols. Further, 15 synthetic derivatives were prepared from these two lead compounds. These compounds (natural and synthetic) were evaluated for their anti-TB activity against M. tuberculosis H37RV. The distinct structure–activity correlations were elucidated based on these derivatives. Derivative 8 showed equivalent activities to those of the compound 1, and it displayed improved anti-TB activity as compared with its parent compound (2). As 8 is a nitrogen-containing compound, it can be made as a water soluble salt, which is considered as valuable in drug development for the improvement of bioavailability. The compound should be regarded as a lead compound for synthesis of additional resorcinol derivatives in the search of novel anti-TB agents.
Acknowledgments
The work described in this article was supported by grants from the Hong Kong Baptist University (HKBU) Interdisciplinary Research Matching Scheme (RC-IRMS/12-13/03), Natural Science Foundation of China (No. 21402166), NIH Grants 3U01TW001015-10S1 and 2U01TW001015-11A1 (administered by the Fogarty International Center as part of an International Cooperative Biodiversity Groups program, through funds from the NIH, NSF, and Foreign Agricultural Service of the USDA), and Shenzhen strategic emerging industry development project funding [CXZZ20150601110000604; ZDSYS201506031617582; Shenfagai (2013) 180].
Author Contributions
Dr. Yi-Fu Guan, Mr. Xun Song, and Dr. Ming-Hua Qiu performed most of the chemistry related experiments including separation, structure determination, and chemical synthesis of the reported compounds with support of Dr. Hong-Jie Zhang, Dr. Harry H.S. Fong, and Dr. Zhen-Dan He. Dr. Shi-Hong Luo did NMR measurement of the synthetic compounds with support of Dr. Sheng-Hong Li. Dr. Nguyen Van Hung performed the extraction of the plant sample. Dr. D. Doel Soejarto and Nguyen Man Cuong collected and authenticated the plant materials. Dr. Bao-Jie Wang performed most of the biology-related experiments including anti-TB evaluation with support of Dr. Scott G. Franzblau. Dr. Hong-Jie Zhang, Dr. Harry H.S. Fong, Dr. D. Doel Soejarto, and Dr. Scott G. Franzblau designed the bioassay-guided separation study. Dr. Hong-Jie Zhang designed the synthetic study. Dr. Yi-Fu Guan, Mr. Xun Song, Dr. Zheng-Dan He, and Dr. Hong-Jie Zhang cowrote the manuscript with the assistance of Dr. Harry H.S. Fong, Dr. Scott Franzblau, and Dr. D. Doel Soejarto. All authors discussed the results and commented on the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.

