

The emergence of the tetrathionate reductase operon in the Escherichia coli/Shigella pan-genome
Graphical Abstract
In recent years, the severity of some outbreaks of food-borne illness ascribed to Escherichia coli has been recognized as due to enhanced virulence brought about by altered genetic characteristics of the implicated pathovars. Herein, we report the isolation of a strain of E. coli possessing a virulence factor normally associated with gastrointestinal infections by Salmonella or Citrobacter. We further provide a detailed examination of the extent and distribution of the tetrathionate reductase genes within strains of E. coli, Shigella flexneri, and Shigella sonnei deposited in Genbank.

Abstract
Escherichia coli pathogenic variants (pathovars) are generally characterized by defined virulence traits and are susceptible to the evolution of hybridized identities due to the considerable plasticity of the E. coli genome. We have isolated a strain from a purified diet intended for research animals that further demonstrates the ability of E. coli to acquire novel genetic elements leading potentially to emergent new pathovars. Utilizing next generation sequencing to obtain a whole genome profile, we report an atypical strain of E. coli, EcoFA807-17, possessing a tetrathionate reductase (ttr) operon, which enables the utilization of tetrathionate as an electron acceptor, thus facilitating respiration in anaerobic environments such as the mammalian gut. The ttr operon is a potent virulence factor for several enteric pathogens, most prominently Salmonella enterica. However, the presence of chromosomally integrated tetrathionate reductase genes does not appear to have been previously reported in wild-type E. coli or Shigella. Accordingly, it is possible that the appearance of this virulence factor may signal the evolution of new mechanisms of pathogenicity in E. coli and Shigella and may potentially alter the effectiveness of existing assays using tetrathionate reductase as a unique marker for the detection of Salmonella enterica.
1 INTRODUCTION
Certain prokaryotes are capable of metabolizing dietary sulfur into hydrogen sulfide (H2S) which can be toxic to mammalian hosts (Nguyen et al., 2020). As part of a remedial mechanism for the detoxification of hydrogen sulfide, the mammalian cecal mucosa converts H2S to thiosulfate (Furne et al., 2001). Subsequently, thiosulfate can then be oxidized by these bacteria to tetrathionate (S4O6²¯) (Barton et al., 2017).
The ability to conduct anaerobic respiration using tetrathionate as an electron acceptor by members of the Enterobacteriaceae family is most commonly found in the genera Salmonella, Proteus, and Citrobacter (Barrett & Clark, 1987). These genera are extensively populated with obligate or opportunistic gastrointestinal pathogens. Their close relative, the species Escherichia coli, is a well-characterized fecal coliform usually acting as a commensal but is also capable of causing intestinal and/or extraintestinal disease. Intestinal pathogenic E. coli strains can be divided into six separate categories or pathotypes including enteroaggregative E. coli (EAEC), entero-invasive E. coli (EIEC), enteropathogenic E. coli (EPEC), enterotoxigenic E. coli (ETEC), enterohaemorrhagic E. coli (EHEC) and diffuse adhering E. coli (DAEC) (Chaudhuri et al., 2010). These E. coli pathovars share a core genome of approximately 2,000 genes. In the continuing evolution of E. coli, the core genomes are augmented by the nonhereditary acquisition of different genes of introgressive origin (Bapteste et al., 2012). As a consequence of the genomic acquisition, a variety of strains emerge possessing an array of adaptive or pathogenic traits (Baquero & Tobes, 2013), thereby displaying a remarkable genomic fluidity (Brunder & Karch, 2000; Hazen et al., 2017; Pasqua et al., 2017; Prager et al., 2014). Perhaps a rather dramatic example of this is the major outbreak caused by Escherichia coli of serotype O104:H4 which spread throughout Europe in 2011. This event, regarded as the most lethal of its kind ever reported (Sloup et al., 2016), was caused by an atypical strain that is most similar to enteroaggregative E. coli (EAEC) of serotype O104:H4. This EAEC variant, however, was found to possess a prophage encoding the Shiga toxin, which is characteristic of enterohemorrhagic E. coli (EHEC) strains. This combination of genomic features, uniting virulence factors from both EAEC and EHEC, represents an example of an emergent new pathovar (Navarro-Garcia, 2014) designated as Enteroaggregative Enterohemorrhagic E. coli, or EAHEC. Introgressive descent refers to “the incorporation (usually via hybridization and backcrossing) of alleles from one entity (species) into the gene pool of a second, divergent entity (species)” (Harrison & Larson, 2014). In this report, we demonstrate a further example of this concept with the discovery of a strain possessing genetic elements previously unreported in E. coli.
Our laboratory routinely tests research animal feeds used at our institute for microbial burden and food-borne pathogens such as Salmonella. While screening a purified, high-fat rodent diet, we isolated a novel strain of E. coli designated EcoFA807-17, displaying an indeterminate biochemical profile while conducting an enrichment protocol for the detection of Salmonella enterica. S. enterica shares a morphological resemblance with EcoFA807-17 on Brilliant Green (Figure 1) and MacConkey Agars, which is also remarkable in the case of Brilliant Green because this selective plate medium tends to inhibit the culture of many strains of E. coli (Moats & Kinner, 1974) including Shigellae (Kristensen, 1925; Moats & Kinner, 1974). After initially characterizing the isolate via multi-locus sequence analysis (MLSA) as E. coli rather than Salmonella, and upon completion of whole genome sequencing (WGS), the isolate appears to possess certain known virulence genes and phenotypes suggesting pathogenic potential, including a functional tetrathionate reductase operon. Tetrathionate respiration, conducted under conditions of anaerobiosis and gut inflammation, serves to enable ttr(+) prokaryotes to outcompete the endogenous intestinal microbiome and is therefore considered a virulence marker in certain enteric pathogens, most notably Salmonella enterica (Winter & Bäumler, 2011).

2 METHODS AND MATERIALS
2.1 Isolation and morphologic/biochemical phenotyping of EcoFA807-17
EcoFA807-17 was initially isolated from culture derived from Rappaport-Vassiliadis broth, then subcultured upon MacConkey (MAC) and Brilliant Green (BG) agars. To initially confirm/disprove Salmonella with which the isolate shared a morphological resemblance on both of these two differential agars, cultures were grown on Triple Sugar Iron (TSI) slant media testing for H2S production (Binet et al., 2018). Additional metabolic phenotyping was performed on the isolate using the Analytic Profile Index (API) 20E system (bioMérieux, Inc.). To differentiate between E. coli and Shigella, isolates were inoculated into Motility Media with Tetrazolium growth Indicator (Hardy Diagnostics). Isolates were also cultured on Xylose-Lysine-Deoxycholate (XLD) Agar, specifically to confirm the isolate's inability to metabolize Xylose (Silva et al., 1980).
2.2 Antibiotic resistance profile analysis
Testing via the disk diffusion method was conducted to determine the susceptibility of EcoFA807-17 to selected members of classes of antimicrobial agents including the beta-lactam, macrolide, fluoroquinolone, aminoglycoside, sulfonamide, tetracycline, and chloramphenicol families.
2.3 Species identification using multi-locus sequence analysis
Multi-locus sequence analysis (MLSA) was performed as part of the initial species identification process. Total genomic DNA (gDNA) was isolated from bacterial colonies using a Qiagen DNeasy Blood and Tissue Kit (Qiagen). Purified extracted genomic DNA (gDNA) was quantified for concentration with a DeNovix DS-11 series Spectrophotometer (DeNovix Inc.). One nanogram (1 ng) of gDNA was used for each Polymerase Chain Reaction (PCR) assay. Bacterial household target genes included 16S rRNA (rrn), β subunit of the RNA Polymerase (rpoB), and DNA gyrase subunit B (gyrB). Gene-specific PCR primers, developed for routine use in our group and proven against E. coli ATCC strain 10536, and the thermocycler parameters for these targets are listed in Table 1. PCR amplicons were purified using the Qiaquick PCR Purification Kit (Qiagen, Co.) and submitted for Sanger sequencing using the same primers (Table 1) (Genewiz, Inc.), and the sequences were analyzed using the CLC Main Workbench software (version 8.1, Qiagen Bioinformatics, Inc.). Sequence alignment and identification (Table 4) were performed using the Basic Local Alignment Search Tool (BLAST; https://blast.ncbi.nlm.nih.gov/Blast.cgi).
| Target gene | Forward | Reverse |
|---|---|---|
| 16S rRNA | 5′-CGGACGGGTGAGTAATGTCT-3′ | 5′-TCAACAACCGAGCTGACGAC-3′ |
| 95°C for 1 min followed by 35 cycles of 95°C for 30 s, 56°C for 30 s, and 72°C for 30 s, and a final extension | ||
| extension step of 72 C for 5 min | ||
| rpoB | 5′-AGACCGTTTCACCATCC-3′ | 5′-ACCCTTGTTACGTGACGAC-3′ |
| 94°C for 1 min followed by 35 cycles of 94°C for 30 s, 55°C for 30 s, and 72°C for 30 s, and a final extension | ||
| step of 72 C for 5 min | ||
| gyrB | 5′-GCGTAACCCGGGTATGTA-3′ | 5′-CCGTCGACGTCCGCATCGGTCAT-3′ |
| 94°C for 1 min followed by 35 cycles of 94°C for 30 s, 60°C for 30 s, and 72°C for 30 s, and a final extension | ||
| step of 72 C for 5 min | ||
| yybW (region 401-611) | 5′-TGATTGGCAAAATCTGGCCG-3′ | 5′-GAAATCGCCCAAATCGCCAT-3′ |
| 95°C for 3 min followed by 40 cycles of 95°C for 15 s, 62°C for 30 s, and 72°C for 30 s. | ||
| Melt Curve 65.0°C to 95.0°C: Increment 0.5°C, 0:05 Plate Read | ||
- Abbreviations: MLSA, multi-locus sequence analysis; PCR, polymerase chain reaction. yybW primer sequences and PCR cycle parameters from Walker et al. (2017).
2.4 Amplification of the E. coli-specific ybbw gene
PCR was performed by targeting the species-defining E. coli ybbW gene as described (primer sequences and PCR parameters are listed in Table 1) (Walker et al., 2017).
2.5 Whole genome sequencing of isolate
Genomic DNA of EcoFA807-17 with a concentration in excess of 4 nM was prepared via Illumina Nextera NT DNA Library Kit (Illumina). DNA sequence data for EcoFA807-17, 5,425,374 million 151 bp paired-end reads, was generated on the Illumina MiSeq platform in the Epigenomics and DNA Sequencing Core at NIEHS. Quality was checked with the Fastqc program (https://bioinformatics.babraham.ac.uk/projects/fastqc/) and no trimming or adaptor removal was indicated. Sequence assemblies and genome annotation were done with Patric 3.5.30 (https://www.patricbrc.org/; (Wattam et al., 2017). Assembly with this web-based toolkit is done with the SPAdes assembler (Bankevich, Nurk, et al., 2012) and resulted in an assembly of 259 contigs. The genome size is 4,964,044 bp and the genome coverage is 330X. Annotation is based on the RASTtk annotation pipeline (Brettin et al., 2015). The annotation advantage of using Patric is that it contains seven databases related to proteins involved in bacterial pathogenesis, virulence, antibiotic resistance, and drug targets. Based on our comparison to prokka (Seemann, 2014), a standard prokaryotic annotation tool, Patric provides a much more accurate annotation of draft prokaryotic genomes and allowed us to identify the ttr operon.
2.6 Analysis of genes involved in tetrathionate respiration
Whole genome sequencing identified genes involved in tetrathionate respiration within EcoFA807-17. To confirm their presence, we first performed PCR analysis utilizing the assay previously reported to detect the Salmonella-specific ttr locus (Malorny et al., 2004). In addition, we performed PCR amplification of the individual ttr genes (A, B, C, R, and S) using PCR primers designed (Table 2) from the ttrRSBCA sequences computationally described in the annotated whole genome sequence data. To confirm the computational prediction of existing ttr genes, partial sequence amplicons of all five genes were successfully generated via PCR (Figure 3) and submitted for sequencing (Genewiz, Inc.), then examined for confirmation of protein function/identification and taxonomic data via the NCBI BLASTX and BLASTN Bioinformatics search tools, respectively.
| Gene | Forward | Reverse |
|---|---|---|
| ttrA | 5′-ACAGCCCGCTACGTATTCTG-3′ | 5′-GCGGGAGTAGATAAACGCCA-3′ |
| ttrB | 5′-CGAAGGCTCACAACAGCATC-3′ | 5′-CACGTCCCATCAATGGGGTA-3′ |
| ttrC | 5′-GTTACCCTGGGCCGTACAAT-3′ | 5′-GTAACGTCCAGCGCATTAGC-3′ |
| ttrR | 5′-GGCCTGTGCGTTTTTATTGGA-3′ | 5′-TTTTGCTACCAGATGCGCCA-3′ |
| ttrS | 5′-TGTTTAACCAGTACGCCGCT-3′ | 5′-ATGGCCAGTCCTAGTCCCAT-3′ |
- Note: The thermal cycling parameters were 95°C for 3 min followed by 35 cycles of 95°C for 15 s for denaturation, 60/62/64°C (60°C - ttrA, ttrC, ttrS, 62C - ttrB, 64C- ttrR) for 30 s for the annealing step, and 72°C for 1 min for extension. A final extension step of 72°C for 5 min was included.
During Polymerase Chain Reaction (PCR) analysis of genes involved in tetrathionate respiration, E. coli (ATCC 10536) was utilized as a ttr(−) negative control. Salmonella enterica (ATCC 13076) and Citrobacter freundii (ATCC 8090) also served as positive controls as both possess a complete ttr operon.
2.7 Growth of EcoFA807-17 in tetrathionate broth
Cultures of EcoFA807-17 and Salmonella enterica, Citrobacter freundii, and ttr(−) E. coli (ATCC 10536) were grown for at least 24 h at 37°C on MacConkey agar. Individual colonies of all species were picked via 1 μl disposable loops to inoculate 16 × 150 ml screw-top tubes containing 10 ml of tetrathionate broth (Hardy Diagnostics). One set of tubes containing all four species was amended with the addition of 200 μl of iodine-iodide solution (Hardy Diagnostics) just before inoculation. A second corresponding set of tubes was inoculated with one of all four species without an amendment of the iodine-iodide solution. All sets were then placed in Gas-Pak sealed jars with CO2 generator packs (Becton Dickinson) to establish an anaerobic growth environment and incubated at 37°C. After 8 h, all tubes of both sets were removed and 10 μl of media from each tube was deposited on MacConkey agar plates, streaked for isolation, and placed in an aerobic incubator for an additional 8 h at 37°C.
3 RESULTS
3.1 Biochemical phenotype and antibiotic resistance profiles of EcoFA807-17
Biochemical profile data of EcoFA807-17 for purposes of taxonomic determination is recorded in Table 3 (with commensal reference strain E. coli ATCC 10536) were derived in part from the Analytic Profile Index (API) 20E system (see Methods and Materials) and from differential media including TSI slant agar for sugar utilization and H2S production, XLD agar for Xylose utilization, and semi-solid Motility media.
| Biochemical test | EcoFA807-17 | E. coli ATCC 10536 |
|---|---|---|
| ONPG | − | + |
| Arginine decarboxylation | − | + |
| Lysine decarboxylation | − | + |
| Ornithine decarboxylation | − | + |
| Citrate utilization | − | − |
| Hydrogen sulfide production | − | − |
| Urease | − | − |
| Tryptophan deaminase | − | − |
| Indole | + | + |
| Vogues–Proskauer | − | − |
| Gelatinase | − | − |
| Glucose | + | + |
| Mannose | + | + |
| Inositol | − | − |
| Sorbitol | + | + |
| Rhamnose | + | + |
| Sucrose | − | + |
| Melibiose | + | + |
| Amygdalin | − | − |
| Arabinose | + | + |
| Lactose | − | + |
| Xylose | − | + |
| Triple sugar iron test | Alkaline/acid/slight gas | Acid/acid/gas |
| Motility | Motile | Motile |
Apart from tetrathionate respiration, the metabolic profile of EcoFA807-17 differs from commensal strains of E. coli in several ways. It does not decarboxylate lysine or ornithine which are known virulence factors among Escherichia/Shigella genomospecies (Casalino et al., 2003; Gomig et al., 2015; Maurelli et al., 1998), although it demonstrates Indole production, a trait long known to be typical of E. coli but much less common with Shigella (Rezwan et al., 2004) and certain other members of Enterobacteriaceae. Its sugar utilization resembles Shigella in being unable to metabolize lactose, sucrose, and xylose (Taylor, 1965). However, EcoFA807-17 differs from Shigella spp. in being robustly motile (Beld & Reubsaet, 2012). In contrast to many pathogens in which wide-spectrum antibiotic resistance is frequently characteristic, EcoFA807-17 was found to be generally susceptible to a broad array of antibiotic agents (Table 4).
| Antibiotic/antibiotic class | Diameters of inhibition zones (mm) | MIC results |
|---|---|---|
| Ampicillin 10 µg/beta-lactams | 24 | Susceptible |
| Azithromycin 15 µg/macrolides | 30 | Susceptible |
| Aztreonam 30 µg/beta-lactams | 39 | Susceptible |
| Cephalothin 30 µg/beta-lactams | 27 | Susceptible |
| Ciprofloxacin 5 µg/fluoroquinolones | 46 | Susceptible |
| Chloramphenicol 30 µg/chloramphenicol | 34 | Susceptible |
| Gentamicin 10 µg/aminoglycosides | 28 | Susceptible |
| Kanamycin 30 µg/aminoglycosides | 22 | Susceptible |
| Meropenem 10 µg/beta-lactams | 37 | Susceptible |
| Sulfamethoxazole/trimethoprim 1.25 µg/sulfonamides | 32 | Susceptible |
| Tetracycline 30 µg/tetracyclines | 25 | Susceptible |
| Tigecycline 15 µg/glycylcyclines (derived from tetracyclines) | 29 | Susceptible |
- Note: Used Mueller–Hinton agar.
- Abbreviation: MIC, minimum inhibitory concentration.
When cultured under anaerobic conditions in (Iodine-Iodide) activated tetrathionate broth to simulate an environment of inflammation within the gut, EcoFA807-17 robustly demonstrated the biological activity of its suite of ttr genes with growth similar to the positive control Salmonella and Citrobacter strains. Conversely, the ttr(−) strain E. coli remained inhibited and failed to grow at the same rate as EcoFA817-17 and the other ttr(+) species when transferred to MacConkey agar (Figure 2). This result was further emphasized with the successful culture of ttr(−) E. coli on MacConkey agar derived from tetrathionate broth that had not been amended with the iodine-iodide solution as seen in Figure 2.

3.2 Multi-locus sequence analysis of EcoFA807-17
The housekeeping gene targets in Table 5 were examined via NCBI BLAST for sequence similarity, confirming EcoFA807-17 as a member of the E. coli/Shigella genomospecies.
| Target gene | Leading NCBI BLAST matches to EcoFA807-17 | Nucleotide identity (%) |
|---|---|---|
| 16S (rrn) | S. sonnei strain ECSW + 14 | 99.77 |
| E. coli strain K-12, sub-strain MG1655 | 99.77 | |
| E. coli O157:H7 strain Sakai | 99.77 | |
| rpoB | E. coli O157:H7 strain Sakai | 99.70 |
| Shigella sonnei strain ECSW + 14 | 99.26 | |
| E. coli strain K-12, sub-strain MG1655 | 99.26 | |
| gyrB | Shigella sonnei strain ECSW + 14 | 99.05 |
| E. coli strain K-12, sub-strain MG1655 | 99.05 | |
| Shigella dysenteriae strain BCW_4872 | 98.84 |
To further differentiate between E. coli and Shigella, the presence of a ybbW (allantoin permease) allele was affirmed by rtPCR assay and by computational prediction derived from whole genome sequencing.
3.3 Sequencing analysis of ttr elements
To confirm the computational prediction of existing ttr genes, partial amplicons of all five genes were created via PCR and submitted for sequencing (Genewiz, Inc.), then examined for confirmation of protein identification and taxonomic data via the NCBI BLASTX and BLASTN Bioinformatics search tools, respectively. Despite the significant divergence in nucleotide identity between the EcoFA807-17 and C. freundii homologs, we were consistently successful in amplifying most of the ttr elements of both species with primers specifically targeting EcoFA807-17 ttr homologs, except for the ttrB structural gene of C. freundii (Figure 3, Lane 15), using stringent annealing temperatures consistent with primer melting temperatures (Tm). By contrast, we were unable to amplify Salmonella ttr homologs, nor did we detect the presence of ttr elements in EcoFA807-17 via a Salmonella-specific PCR assay (Malorny et al., 2004), thus providing additional support for the possibility that EcoFA807-17 has acquired its ttr elements from a Citrobacter lineage.

The PCR amplicons of the various ttr locus elements were resolved on a 2.0% agarose gel. The lanes are as follows:
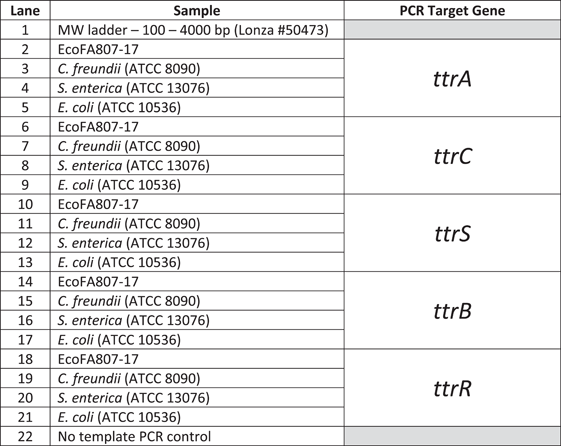
Note: Trace bands in Lanes 16, 20, and 21 were subjected to additional PCR and determined via sequencing and BLAST analysis to be nonspecific amplification.
3.3.1 Distribution of the ttr operon in E. coli strains available at NCBI
All 20833 E. coli genomes listed in the E. coli Genome Assembly and Annotation Report (https://www.ncbi.nlm.nih.gov/genome/genomes/167/) were downloaded on 22 September 2020, and searched for those potentially containing genes in the ttr operon using the proteins of the ttr operon of EcoFA807-17 as queries. The number of strains containing each of the five genes of this operon and the subset of that containing all five genes of the ttr operon are listed in Table 6. Only approximately 0.7% of these genomes contained all five genes comprising the ttr operon, whereas the lacZ gene common to coliform bacteria (Molina et al., 2015), and the ybbW gene, diagnostic of E. coli, were present in 97.8 and 96.3 of this E. coli genome data set, respectively.
| Gene | Genomes containing gene | Percent of total genomes with gene |
|---|---|---|
| ttrR | 166 | 0.79 |
| ttrS | 166 | 0.79 |
| ttrB | 165 | 0.79 |
| ttrC | 167 | 0.80 |
| ttrA | 163 | 0.78 |
| ttr operon | 146 | 0.70 |
| lacZ | 20,380 | 97.80 |
| ybbW | 20,075 | 96.30 |
To confirm that these genes were present in a true operon configuration, EcoFA807-17 and a sample of 27 complete E. coli genomes of those strains in Table 7 predicted to have a complete ttr operon were re-annotated using the Patric annotation pipeline described in Methods. Where the assembly annotation indicated a plasmid localization of the ttr operon is also noted in Table 7.
| Strain possessing complete ttr operon | Date submitted to NCBI | Accession | Operon locus |
|---|---|---|---|
| 2.4168 | 2-May-12 | GCA_000194555.2 | Chromosome |
| 541-15 | 23-May-12 | GCA_000264115.1 | Chromosome |
| KTE111 | 5-Apr-13 | GCA_000351865.1 | Chromosome |
| KTE40 | 4-Jun-13 | GCA_000408025.1 | Chromosome |
| KTE64 | 4-Jun-13 | GCA_000408365.1 | Chromosome |
| UMEA 3052-1 | 29-Aug-13 | GCA_000460035.1 | Chromosome |
| UMEA 3889-1 | 29-Aug-13 | GCA_000461755.1 | Chromosome |
| UMEA 3148-1 | 1-Nov-13 | GCA_000495075.1 | Chromosome |
| D6-117.29 | 11-May-14 | GCA_000723325.1 | Chromosome |
| 2-156-04_S4_C2 | 11-Jun-14 | GCA_000704025.1 | Chromosome |
| 2-474-04_S1_C1 | 11-Jun-14 | GCA_000704565.1 | Chromosome |
| 2-474-04_S1_C2 | 11-Jun-14 | GCA_000704645.1 | Chromosome |
| CVM N41498PS | 7-Dec-14 | GCA_000797915.1 | Chromosome |
| 8.0569 | 18-Mar-15 | GCA_000305355.1 | Chromosome |
| YE16 | 7-Aug-15 | CXWW01000045.1 | Chromosome |
| YE19 | 7-Aug-15 | CXYJ01000044.1 | Chromosome |
| AW1.7 1161 | 9-Oct-15 | GCA_001309455.1 | Chromosome |
| AW1.7-delta-pHR1 996 | 9-Oct-15 | GCA_001309475.1 | Chromosome |
| GM16-6 | 9-Oct-15 | GCA_001309555.1 | Chromosome |
| RU1 | 26-Oct-15 | GCA_001413005.1 | Chromosome |
| RU1 MA1 | 26-Oct-15 | GCA_001413355.1 | Chromosome |
| RU1 MA3 | 26-Oct-15 | GCA_001413395.1 | Chromosome |
| RU1 MA4 | 26-Oct-15 | GCA_001413415.1 | Chromosome |
| RU1 MA5 | 26-Oct-15 | GCA_001413425.1 | Chromosome |
| RU1 MA6 | 26-Oct-15 | GCA_001412895.1 | Chromosome |
| RU1 MA7 | 26-Oct-15 | GCA_001413455.1 | Chromosome |
| RU1 MA8 | 26-Oct-15 | GCA_001412915.1 | Chromosome |
| RU1 MA9 | 26-Oct-15 | GCA_001413475.1 | Chromosome |
| RU1 MA10 | 26-Oct-15 | GCA_001413485.1 | Chromosome |
| RU1 MA11 | 26-Oct-15 | GCA_001413515.1 | Chromosome |
| RU1 MA12 | 26-Oct-15 | GCA_001413535.1 | Chromosome |
| RU1 MA12 | 26-Oct-15 | GCA_001413535.1 | Chromosome |
| RU1 LB1 | 26-Oct-15 | GCA_001412925.1 | Chromosome |
| RU1 LB2 | 26-Oct-15 | GCA_001412955.1 | Chromosome |
| RU1 LB3 | 26-Oct-15 | GCA_001413555.1 | Chromosome |
| RU1 LB4 | 26-Oct-15 | GCA_001413565.1 | Chromosome |
| RU1 LB5 | 26-Oct-15 | GCA_001412975.1 | Chromosome |
| RU1 LB6 | 26-Oct-15 | GCA_001413595.1 | Chromosome |
| RU1 LB7 | 26-Oct-15 | GCA_001413605.1 | Chromosome |
| RU1 LB9 | 26-Oct-15 | GCA_001412995.1 | Chromosome |
| RU1 LB10 | 26-Oct-15 | GCA_001413645.1 | Chromosome |
| RU1 LB11 | 26-Oct-15 | GCA_001413675.1 | Chromosome |
| RU1 LB12 | 26-Oct-15 | GCA_001413685.1 | Chromosome |
| RU1 BHI2 | 26-Oct-15 | GCA_001413735.1 | Chromosome |
| RU1 BHI3 | 26-Oct-15 | GCA_001413745.1 | Chromosome |
| RU1 BHI4 | 26-Oct-15 | GCA_001413755.1 | Chromosome |
| RU1 BHI5 | 26-Oct-15 | GCA_001413795.1 | Chromosome |
| RU1 BHI7 | 26-Oct-15 | GCA_001413875.1 | Chromosome |
| RU1 BHI9 | 26-Oct-15 | GCA_001413885.1 | Chromosome |
| RU1 BHI11 | 26-Oct-15 | GCA_001413905.1 | Chromosome |
| RU1 BHI12 | 26-Oct-15 | GCA_001413825.1 | Chromosome |
| AZ71 | 4-Jan-16 | GCA_001484375.1 | Chromosome |
| G138 | 26-Feb-16 | GCA_001575835.1 | Chromosome |
| G150 | 26-Feb-16 | GCA_001575865.1 | Chromosome |
| G184 | 26-Feb-16 | GCA_001575945.1 | Chromosome |
| G186 | 26-Feb-16 | GCA_001576295.1 | Chromosome |
| G228 | 26-Feb-16 | GCA_001575705.1 | Chromosome |
| G239 | 26-Feb-16 | GCA_001575695.1 | Chromosome |
| G240 | 26-Feb-16 | GCA_001575635.1 | Chromosome |
| G3 | 26-Feb-16 | GCA_009823265.1 | Chromosome |
| G35 | 26-Feb-16 | GCA_001576385.1 | Chromosome |
| AF7945 | 29-Sep-16 | GCA_001749075.1 | Chromosome |
| 639 | 8-Dec-16 | GCA_001893305.1 | Chromosome |
| 643 | 8-Dec-16 | GCA_001893215.1 | Chromosome |
| 491 | 8-Dec-16 | GCA_001893375.1 | Chromosome |
| 552 | 8-Dec-16 | GCA_001893225.1 | Chromosome |
| 584 | 8-Dec-16 | GCA_001894315.1 | Chromosome |
| 602 | 8-Dec-16 | GCA_001893995.1 | Chromosome |
| 647 | 8-Dec-16 | GCA_001894425.1 | Chromosome |
| 684 | 8-Dec-16 | GCA_001893765.1 | Chromosome |
| H15 | 12-Dec-16 | GCA_001901005.1 | Plasmid |
| 299.h | 1-Sep-17 | GCA_002284735.1 | Chromosome |
| F1_405E | 6-Sep-17 | GCA_900195635.1 | Chromosome |
| MOD1-EC3803 | 5-Oct-17 | GCA_002516065.1 | Chromosome |
| MOD1-EC3800 | 5-Oct-17 | GCA_002516125.1 | Chromosome |
| MOD1-EC6602 | 6-Oct-17 | GCA_002467595.1 | Chromosome |
| MOD1-EC5157 | 6-Oct-17 | GCA_002516885.1 | Chromosome |
| MOD1-EC6855 | 11-Oct-17 | GCA_002520525.1 | Chromosome |
| MOD1-EC6132 | 13-Oct-17 | GCA_002537315.1 | Chromosome |
| MOD1-EC6011 | 13-Oct-17 | GCA_002542995.1 | Chromosome |
| MOD1-EC6007 | 13-Oct-17 | GCA_002538315.1 | Chromosome |
| MOD1-EC5984 | 13-Oct-17 | GCA_002543795.1 | Chromosome |
| YH17167 | 25-Feb-18 | GCA_002941405.1 | Chromosome |
| EBJ001 | 3-Mar-18 | GCA_002967885.1 | Chromosome |
| B29595 | 8-Mar-18 | GCA_003007795.1 | Chromosome |
| KG-3 | 11-Mar-18 | GCA_002993705.1 | Chromosome |
| 13561-5 | 31-Mar-18 | GCA_003027475.1 | Chromosome |
| KG-18 | 10-Apr-18 | GCA_003048005.1 | Chromosome |
| Win2012_WWKa_NEU_31 | 21-May-18 | GCA_003145195.1 | Chromosome |
| Sum2013_WWKa_OUT_2 | 21-May-18 | GCA_003145395.1 | Chromosome |
| Spr2013_WWKa_ALT_27 | 21-May-18 | GCA_003145945.1 | Chromosome |
| Spr2012_WWKa_NEU_74 | 21-May-18 | GCA_003146135.1 | Chromosome |
| TUM15671 | 9-Jun-18 | GCA_003227915.1 | Chromosome |
| VREC0596 | 18-Jun-18 | GCA_900480485.1 | Chromosome |
| VREC0525 | 18-Jun-18 | GCA_900481835.1 | Chromosome |
| VREC0631 | 18-Jun-18 | GCA_900482185.1 | Chromosome |
| A24 | 9-Jul-18 | GCA_003292515.1 | Chromosome |
| A39 | 9-Jul-18 | GCA_003292875.1 | Chromosome |
| 1262 | 9-Jul-18 | GCA_003293115.1 | Chromosome |
| A48 | 9-Jul-18 | GCA_003293255.1 | Chromosome |
| 1257 | 9-Jul-18 | GCA_003301735.1 | Chromosome |
| JL39 | 9-Jul-18 | GCA_003291015.1 | Chromosome |
| GER_MD01_1509_Eco_058 | 15-Jul-18 | GCA_003322075.1 | Chromosome |
| GER_MD11_1505_Eco_029 | 15-Jul-18 | GCA_003322395.1 | Chromosome |
| GER_MD11_1505_Eco_027 | 15-Jul-18 | GCA_003322415.1 | Chromosome |
| GER_MD11_1505_Eco_023 | 15-Jul-18 | GCA_003322465.1 | Chromosome |
| KL53 | 20-Jul-18 | GCA_002494365.2 | Plasmid |
| NCTC11130 | 30-Jul-18 | GCA_900449175.1 | Chromosome |
| NCTC13384 | 31-Jul-18 | GCA_900448435.1 | Chromosome |
| NCTC11106 | 31-Jul-18 | GCA_900448865.1 | Chromosome |
| HPC-781c | 20-Sep-18 | GCA_003583685.1 | Chromosome |
| PN112 | 26-Nov-18 | GCA_003830335.1 | Chromosome |
| PN91 | 26-Nov-18 | GCA_003830385.1 | Chromosome |
| GBGD39 | 3-Dec-18 | GCA_003858925.1 | Chromosome |
| NCTC11129 | 19-Dec-18 | GCA_900636075.1 | Chromosome |
| 49_rectal | 21-Jan-19 | GCA_004100615.1 | Chromosome |
| RS571 | 25-Jan-19 | GCA_004114395.1 | Plasmid |
| 071H1 | 6-Feb-19 | GCA_004173925.1 | Chromosome |
| 93-I92-A | 14-Mar-19 | GCA_005402105.1 | Chromosome |
| URMC_46 | 2-Apr-19 | GCA_004567595.1 | Chromosome |
| KCJK7052 | 9-Apr-19 | GCA_004767325.1 | Chromosome |
| BE565 | 16-Apr-19 | GCA_005381805.1 | Chromosome |
| JML241 | 16-Apr-19 | GCA_005388945.1 | Chromosome |
| EC-129 | 6-May-19 | GCA_005156265.1 | Plasmid |
| SCEC020023 | 19-Aug-19 | GCA_002850675.5 | Plasmid |
| CD64_9 | 20-Aug-19 | GCA_008040635.1 | Chromosome |
| 1_53_1 | 20-Aug-19 | GCA_008041255.1 | Chromosome |
| 1_52_13 | 20-Aug-19 | GCA_008041305.1 | Chromosome |
| P042A | 27-Aug-19 | GCA_008120445.1 | Chromosome |
| C27A | 18-Dec-19 | GCA_009762475.1 | Plasmid |
| SH9c | 22-Dec-19 | GCA_009789905.1 | Chromosome |
| IH27c | 22-Dec-19 | GCA_009791155.1 | Chromosome |
| IH8c | 22-Dec-19 | GCA_009791245.1 | Chromosome |
| IH3c | 22-Dec-19 | GCA_009791265.1 | Chromosome |
| G39 | 31-Dec-19 | GCA_001576155.1 | Chromosome |
| 8374wF12 | 14-Jan-20 | GCA_009882595.1 | Chromosome |
| ATCC 11231 | 10-Feb-20 | GCA_010374855.1 | Chromosome |
| ATCC 11229 | 10-Feb-20 | GCA_010374945.1 | Chromosome |
| EC8 | 25-Feb-20 | GCA_011008605.1 | Chromosome |
| CAP45 | 26-Feb-20 | GCA_011028105.1 | Chromosome |
| CAP36 | 26-Feb-20 | GCA_011032125.1 | Chromosome |
| 169757 | 29-Feb-20 | GCA_011043615.1 | Plasmid |
| Fec 10 | 23-Jun-20 | WP_001076427.1 | Plasmid |
| HUM-546 | 13-Jul-20 | GCA_013404135.1 | Chromosome |
| G43 | 17-Jul-20 | GCA_001576495.1 | Chromosome |
| GCPRC7 | 28-Jul-20 | GCA_013850805.1 | Chromosome |
A similar strategy was used to search available NCBI Shigella genomes, as of 5 May 2020. One S. sonnei strain was found to possess the ttr operon, as were just three other additional Shigella strains including one strain of S. flexneri and two Shigella strains of undetermined species. No S. dysenteriae or S. boydii genomes were found to contain a ttr operon.
Shigella strains containing a complete ttr operon are listed in Table 8. Shigella genomes were formatted into a BLAST database and searched for the five protein queries from the ttr operon from EcoFA807-17 by BLAST. Dates of isolation (where available) and NCBI accession numbers are given in columns 3 and 4 of Table 8. One additional strain, Shigella spp. FC1967 (GCA_001743005.1) possessed four of the five genes found in the ttr operon.
| Species | Ttr(+) strain | Accession | Year submitted | ybbW and ttr(+) | ybbW (+) genomes * |
|---|---|---|---|---|---|
| S. sonnei | ESCW+10 | GCA_002248745.1 | 2017 | yes | 30 |
| S. flexneri | 1235-66 | GCA_000268065.1 | 2012 | no | 11 |
| Shigella spp. | FC1655 | GCA_001742985.1 | 2016 | no | NA |
| Shigella spp. | FC130 | GCA_001722135.1 | 2016 | no | NA |
| Shigella spp. | FC1967 | GCA_001743005.1 | 2016 | no | NA |
- Note: (*) number of ybbW (+) Shigella species genomes deposited in GenBank.
4 DISCUSSION
4.1 Taxonomic determination of species
As E. coli had not previously been reported to possess tetrathionate reductase genes (Palumbo & Alford, 1970; Price-Carter et al., 2001; Roth, 2017), confirmation of the taxonomic classification of EcoFA807-17 as E. coli was essential. This strain presents a conflicting and atypical biochemical identity profile, particularly confounding differentiation between E. coli and Shigella. EcoFA807-17 resembles Shigella in some respects rather than E. coli, most strikingly by its inability to ferment xylose (Altwegg et al., 1996; de Boer, 1998; Taylor, 1965). Further complicating differentiation between E. coli and the obligate pathogen Shigella, the close phylogenetic relationship between the two organisms thwarts the use of household genes typically used in multi-locus sequence analysis for identification at the species level. Those MLSA targets used for the initial genetic examination include rrn (ribosomal RNA or 16S), gyrB, and rpoB. The sequencing and attempted species identification via these genes deserve further comment. Long considered the “gold standard” for taxonomic classification (Case et al., 2007) of bacterial species, the rrn (16S) gene is effective for taxonomic determination at the genus level and sometimes at the species level as well, but is unable to decisively discriminate between E. coli and Shigella (Christensen et al., 1998; Devanga Ragupathi et al., 2018), although its employment was initially instrumental in confirming that EcoFA807-17 was not Salmonella. This was not surprising given that Escherichia and Shigella, while deemed separate taxa for purposes of clinical distinction (Farmer et al., 1985; Rezwan et al., 2004), are genetically the same species (Beld & Reubsaet, 2012; Fukushima et al., 2002). They are therefore too closely related to be reliably differentiated by the highly conserved and frequently multi-copy ribosomal RNA genes (Devanga Ragupathi et al., 2018). Similarly, the gyrB and rpoB household genes which are generally more discriminatory than rrn (16S) at the species and even sub-species level, were determined nonetheless to be insufficiently useful to differentiate between E. coli and Shigella as demonstrated by inconclusive results (Table 5) obtained from NCBI data of the housekeeping gene sequences submitted. By extension, they are also deemed wholly unsuitable for serovar/biovar determination (Adékambi et al., 2009).
Accordingly, a real-time PCR assay described by Walker et al., (Walker et al., 2017) was utilized to enable a decisive confirmation of EcoFA807-17 as E. coli. The principle underlying this method is that the ybbW gene, which codes for allantoin permease, has been regarded as universally inclusive and exclusive to E. coli (but not Shigella) and subsequent testing confirmed that EcoFA807-17 is ybbW (+). We observe, however, that some data from this study suggests that ybbW may not be as exclusive to E. coli as originally thought. Specifically, we note that a number of strains of S. sonnei and S. flexneri found within NCBI data (Table 8, right column) appear to possess the ybbW gene, including ttr(+) S. sonnei strain ECSW + 10 (GCA_002248745.1). In the case of the latter, while conceivable that ECSW + 10 is an E. coli (EIEC) strain originally misclassified as S. sonnei, this is a less likely explanation as further examination of the genome of S. sonnei strain ECSW + 10 reveals the predicted presence of a pINV type B invasion plasmid. The latter is characteristic of S. sonnei while closely related EIEC strains and certain other Shigellae tend to possess pINV Type A invasion plasmids (Lan et al., 2001, 2004). The unexpected presence of ybbW as well as a ttr operon within S. sonnei strain ECSW + 10 serves to further emphasize the dynamic mutability of the E. coli/Shigella pan-genome. Nevertheless, the presence of the ybbW gene is observed to be uncommon among the several thousand Shigella genomes deposited at NCBI. For that reason and because the Shigellae universally possess pINV invasion plasmids and lack motility (Beld & Reubsaet, 2012), we conclude that EcoFA807-17, which lacks the former and is highly motile, is properly classified as a strain of E. coli.
4.2 Characterization of the ttr operon of EcoFA807-17
Initially (in 2018), the ttr operon sequences of EcoFA807-17 were determined via NCBI BLASTN to be most closely related to Citrobacter freundii homologs with shared nucleotide identity ranging from 85% to 90% as described from NCBI data. Reinforcing this data, we found that our ttr PCR primers, designed from sequences predicted of EcoFA807-17, not only successfully PCR-amplified and confirmed the computationally indicated existence of these genes, but also PCR-amplified four out of the five counterparts found in C. freundii (see Figure 3), despite the significant degree of deviation in nucleotide identity of the latter species. Recently, a follow-up query to NCBI revealed new sequences had become available, and we have determined that all ttr elements in EcoFA807-17 are much more closely related to homologs found within Citrobacter amalonaticus and to a slightly lesser extent, a novel species Citrobacter portucalensis (Ribeiro et al., 2017), in both cases sharing approximately 99% identity with EcoFA807-17. Of special note with respect to C. portucalensis, due to its recent taxonomic classification, only a few studies relevant to this species have been published and none have reported its possession of ttr genetic elements. As for the uncommon presence of the ttr operon in E. coli, regarding its origin and nearly perfect nucleotide identity with certain homologs within Citrobacter, it is most likely the result of horizontal transfer from a related species. The EcoFA807-17 genome was examined for plasmid content using three tools: plasmidSpades, Recycler (https://github.com/Shamir-Lab/Recycler#bam-prep), and PlasmidSeeker (https://github.com/bioinfo-ut/PlasmidSeeker). Two plasmids predicted by all three tools were considered candidate plasmids. Based on BLAST analysis, the ttr operon locus is not on either of these two plasmids and thus is likely chromosomal. This is consistent with the fact that of the ttr(+) strains listed in Table 7, relatively few appear to possess plasmid-borne ttr elements. To address the question of whether ttr elements within EcoFA807-17 were biologically active, the strain was cultured anaerobically in tetrathionate broth as previously described. Simulation of the in vivo infection and inflammation process is achieved with the addition of the Iodine-iodide solution to the broth. Metabolism of tetrathionate by bacteria imparts a growth advantage under anaerobic conditions and requires the possession of a fully functional ttr operon. Tetrathionate broth has been commonly used for the cultivation of Salmonella specifically because it inhibits growing cells of many ttr(−) Gram-negative species, and in particular, coliforms such as E. coli. As with the Salmonella and Citrobacter positive control strains, EcoFA807-17 was easily cultured in tetrathionate broth, whereas the ttr negative control strain of E. coli remained inhibited, except in tetrathionate broth that had not been oxidized via the addition of iodine-iodide solution.
4.3 Potential origins of the acquisition of ttr operon
We approached the question of origin for the type of ttr operon now established in the E. coli/Shigella pan-genome with an examination of individual ttr gene homology within all known ttr(+) strains among E. coli (146 in total) and an assessment of the syntenic conservation of genetic elements immediately upstream and downstream of the ttr operon chromosomal loci, using EcoFA807-17 and twenty-seven randomly chosen ttr(+) E. coli strains from Table 7. In choosing the ttrA structural gene as representative for the operon, we ascertained that EcoFa807-17 shared 100% nucleotide identity with approximately 51% of strains of ttr(+) E. coli (74/146) and all of the very few known ttr(+) Shigella strains (total of 5). 43% of ttr(+) strains (63/146) shared between 99% and 100% nucleotide identity with EcoFA807-17. The remaining 6% of ttr(+) strains possessed ttr homologs considerably below the threshold of 99% nucleotide shared identity. This is consistent with the fact that this study has earlier determined that ttr homologs within the genus Citrobacter possess varying degrees of sequence conservation as demonstrated by the significant difference between C. freundii and C. amalonaticus. We then further examined the synteny of chromosomal elements in the immediate vicinity of the ttr operon among the same twenty-eight ttr(+) E. coli genomes including EcoFA807-17. A common feature of the surrounding loci revealed the frequent presence of the Methionine ABC transporter substrate binding and Fumarate hydratase genes within ~86% of the E. coli strains sampled, immediately adjacent upstream and downstream respectively of the operon, while every strain possessed at least one of these two flanking genes. Extending this analysis to homologs examined within Citrobacter freundii (ATCC 8090), Citrobacter portucalensis (strain FDAARGOS_617), and Salmonella enterica (ATCC 13076), the same association of the aforementioned adjacent genes is uniformly present in both Citrobacter species, whereas only the Fumarate hydratase gene is associated with the Salmonella ttr operon. Beyond these two operon-flanking genes, however, the degree of conserved synteny among these 28 strains degenerated immediately thereafter. Finally, we further attempted to address the additional question of whether Salmonella ttr genes have perhaps breached the genome of any strain of E. coli or Shigella found in public databases but found that there is insufficient evidence that this has ever occurred. In consideration of these findings, one main conclusion is apparent. Variation in the content of genes in the immediate vicinity of the ttr operon coupled with the presence of nearby mobile genetic elements on either side (insertional sequences, transposases, and assorted others) strongly suggest that acquisition of the ttr operon within the E. coli/Shigella pan-genome is the result of horizontal transfer that has occurred on multiple occasions, with gene homology indicating various Citrobacter species as the most likely point(s) of origin.
4.4 Significance of ttr operon acquisition
There is a relative paucity of literature before this century concerning tetrathionate respiration in the Enterobacteriaceae family since the phenomenon was first described by Pollock and co-workers in the early 1940s (Kapralek, 1972). However, studies conducted in the 1970s by Kapralek and Rezbova with their work confined to the genus Citrobacter, revealed that tetrathionate respiration enhanced specific growth rate, raised growth yield, and enabled growth on non-fermentable carbon sources. By 1999, Hensel and colleagues had identified and mapped the structure and functions of the operon responsible for tetrathionate metabolism in Salmonella (Hensel et al., 1999). Even then, the role of tetrathionate reductase as a virulence factor was not yet established as Hensel surmised that ttr metabolism was a survival trait utilized by Salmonella in the external environment rather than within eukaryotic hosts. This assumption is understandable, given that there were no known sources of tetrathionate in the mammalian host (Winter et al., 2010). Nevertheless, by 2010, Winter, Baumler, and associates had established that by process of induced gut inflammation and under anaerobic conditions, thiosulfate, which is endogenous within mammalian hosts, is oxidized to tetrathionate whereby the latter then becomes a terminal electron acceptor enabling respiration. As Bliska et al. noted, “one of the most important functions of the microbiota of the mammalian intestine is to promote resistance to colonization by pathogens” (Bliska & van der Velden, 2012). Winter et al. demonstrated that Salmonella and presumably other pathogens capable of tetrathionate reduction and respiration, gain a competitive growth advantage over fermentation-dependent gut microbiota in anoxic environments (Winter et al., 2010).
4.5 Pathogenic potential of ttr(+) Escherichia coli
Given the available subject literature, studies of tetrathionate respiration as a virulence factor have generally focused on the obligate food-borne pathogen Salmonella enterica. The genus Citrobacter, many species of which are known to possess the ttr operon, is closely related to Escherichia and it should be re-emphasized that the ttr elements of E. coli strain EcoFA807-17 are much more closely related to Citrobacter in terms of nucleotide identity and significantly more distantly related to the Salmonella ttr operon. Composed of thirteen species accepted by the International Committee on Systematics of Prokaryotes (Ribeiro et al., 2017), some Citrobacter are recognized as opportunistic pathogens such as the ttr(+) species C. freundii and C. amalonaticus (Lipsky et al., 1980). Nevertheless and apart from Salmonella, the literature is sparse concerning the role of tetrathionate respiration as a virulence factor in other bacteria (Hensel et al., 1999) including Citrobacter. Against this backdrop, predictions of ttr-mediated virulence in EcoFA807-17 are confined to the realm of speculation pending further study, even as this novel strain clearly possesses other genetic hallmarks suggesting pathogenicity. These potentially virulent elements include intriguing examples such as eae (Intimin) and prp (Pathogenesis-related Protein), an enigmatic rare gene detected in only nine E. coli genomes obtained from NCBI. Additionally, it is interesting to note that this isolate's Type VI secretion system (T6SS) appears to be of similar lineage to those of Shigella sonnei, st. 53 G and EHEC O157:H7. In the case of the former, it has been recently reported that S. sonnei possesses a T6SS which specifically “competes against E. coli and S. flexneri, both in vitro and in vivo.” (Anderson et al., 2017) As tetrathionate respiration also serves to promote those organisms possessing that capability in interbacterial competition, EcoFA807-17 may potentially have a formidable advantage over endogenous microbiota.
Our data (Table 7) suggests that the operon is gradually proliferating through the past decade in Escherichia coli, though it is present in less than 1% of genomes deposited at NCBI. In contrast, its presence in Shigella (Table 8) is extremely rare, having been found in just five strains among over 2000 genomes examined. In any event, it is now clear that the tetrathionate reductase operon is becoming established within the Escherichia coli/Shigella pan-genome, with future implications yet to be determined.
AUTHOR CONTRIBUTIONS
Floyd G. Adsit: Conceptualization (lead); data curation (lead); formal analysis (lead); investigation (lead); methodology (equal); writing–original draft (lead); writing–review & editing (equal). Thomas A. Randall: Formal analysis (equal); investigation (equal); writing–original draft (equal); writing–review & editing (equal). Jacqueline Locklear: Investigation (supporting); writing–review & editing (supporting). David M. Kurtz: Funding acquisition (lead); resources (equal); supervision (lead); writing–original draft (equal); writing – review & editing (equal).
ACKNOWLEDGMENTS
We wish to acknowledge the invaluable assistance with literature research of Stacey Mantooth, MSLS, staff librarian for the National Institute of Environmental Health Sciences, and Steven R. McCaw, a photographer at the National Institute of Environmental Health Sciences for imagery assistance. This research was supported by the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences.
CONFLICT OF INTEREST
None declared.
ETHICS STATEMENT
None required.
Open Research
DATA AVAILABILITY STATEMENT
In addition to the sequence submission to the Short Read Archive (accession number SRR14216030), this Whole Genome Shotgun project has also been deposited at DDBJ/ENA/GenBank under the accession JAGRQF000000000: The version described in this paper is version JAGRQF010000000: https://www.ncbi.nlm.nih.gov/bioproject/PRJNA721428.

