

Catalytic Enantioselective Functionalization of Maleimides: An Update
Comprehensive Summary
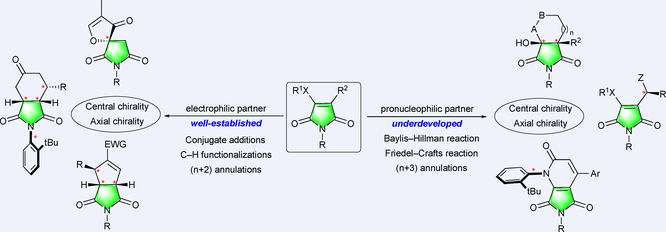
Maleimide derivatives are well-established reactive intermediates also found in natural products, synthetic pharmaceuticals and functional polymers. Their specific reactivity found widespread applications in the field of bioconjugation and allowed for the development of highly selective functionalizations based on simple additions and cycloadditions with the possible control of central and C–N axial chirality. These multisite-reactive scaffolds have aroused a long-standing interest throughout the scientific community more particularly as powerful electrophilic partners and more recently as nucleophilic partners in some specific transformations. The persistent interest in these easily accessible synthetic platforms over the last decade has enabled the development of new enantioselective transformations and the major advancements in this field are presented in this review.
Key Scientists
1 Introduction
When the very first syntheses of maleimides were published in the second part of the 19th century,[1] it was difficult to imagine the growing interest aroused by this new heterocyclic framework over the last 150 years. Nowadays, this peculiar five-membered ring imide core has been identified in various families of natural products,[2] synthetic pharmaceuticals[3] and functional polymers (NSP)[4] offering a wide range of scientific and technological applications.[5] In particular, for more than fifty years, maleimide derivatives have stood out in the field of bioconjugation as privileged partners for the site selective modification of proteins by virtue of their easy accessibility and very good chemical behavior towards nucleophilic biomolecules (Figure 1).[6]

At the same time, the reactivity of multisite-functional maleimides has stimulated a growing interest in the field of synthetic organic chemistry as well-established electrophilic partners for a large panel of nucleophilic conjugate additions,[7] C–H functionalizations[8] and (n+2) cycloadditions.[9] More recently, the underdeveloped unexpected potentially pro-nucleophilic character of maleimide derivatives was brought to light thanks to the development of organocatalysis for the Morita−Baylis−Hillman reaction,[10] the Friedel−Crafts type functionalization[11] and for (n+3) annulations (Figure 2).[12]

Although the enantioselective functionalization of maleimides has been the subject of several specific reviews,[7-9] the persistent interest in this easily available multisite-reactive synthetic platform justifies an update providing the readers of enantioselective catalysis with the major advancements in this field over the last decade. The review is organized in two main sections covering the utilization of maleimides as electrophilic partners (Section 1) and pro-nucleophilic partners (Section 2) in metal-catalyzed or organocatalyzed functionalizations with the control of either central and/or axial chirality.
2 Maleimides as Electrophilic Partners
2.1 Metal catalyzed functionalizations
2.1.1 Nucleophilic additions
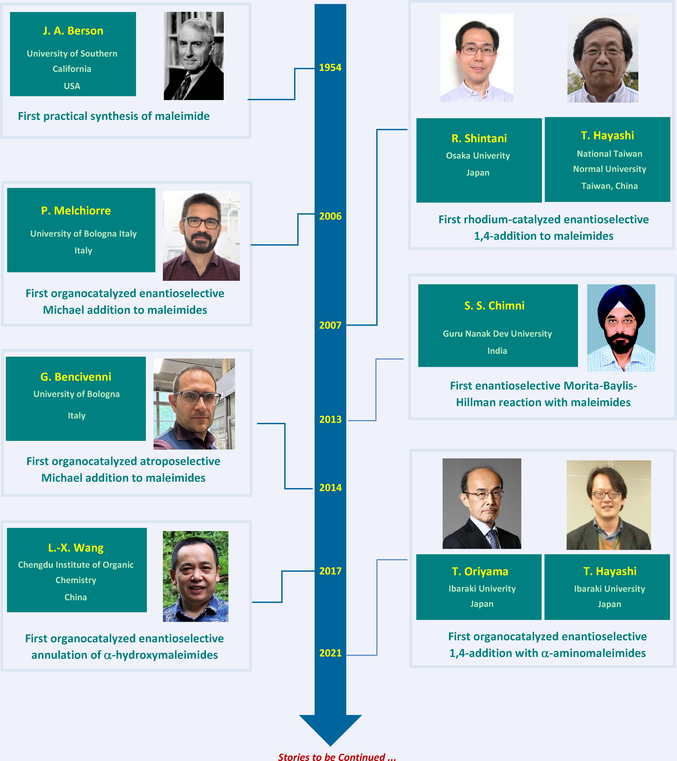
Enantioenriched 3-aryl-succinimides and pyrrolidines have demonstrated to be characteristic cores with a broad spectrum of biological properties, opening new avenues for the development of active drugs and pharmaceutical leads.[13] Thus, considerable attention has been given to their synthetic accessibility and particularly to their enantioselective synthesis as chiral differentiation can dramatically modify the bioactivity of each enantiomer. In this context, the enantioselective 1,4-addition of arylboronic acid derivatives 1 to N-substituted maleimides 2 offers a quite general and efficient strategy to access the desired chiral 3-aryl-succinimides 3a through desymmetrization. So far, several catalytic systems based on rhodium catalysts ligated with various chiral ligands (dienes, sulfoxides, electron-deficient diphosphines, etc.) have been reported with relatively low enantioselectivities, often depending on the nature of the N-substituent of the maleimide substrate.[14] Such a context opens the way to the design and synthesis of novel and more performant catalytic systems. With the aim to improve enantioselectivities, Korenaga's group exploited the beneficial effect of MeO-F12-BIPHEP L1 as electron-deficient phosphorous-containing ligand for the development of low-temperature conditions down to –80 °C (Scheme 1).[15] Reactions were performed between 0 and –50 °C with satisfactory yields and enantioselectivities (87%–99% ee) using various N-substituted maleimides or more interestingly NH-free maleimides (R2 = H). The use of novel chiral 2,5-diaryl substituted bicyclo[2.2.1] dienes such as L2, pioneered by Hayashi,[14] allowed as well to perform under mild conditions the highly enantioselective 1,4-addition of a wide range of arylboronic acids with N-substituted maleimides.[16] Albeit with poor regioselectivity, 1,4-adducts could be generated with high enantioselectivities from N-substituted-3-methylmaleimides (R1 = Me), the reaction occurring preferentially to the less-hindered position delivering 3,4-trans-disubstituted maleimides 3c (Scheme 1).

Although the use of palladium catalysis is less described, a recent achievement has been made by the group of Li by the design of new heterocycle-oxazoline ligands L3 and L4 under relatively mild conditions (Scheme 2).[17] This process offers a large substrate scope and some of the targeted 3-arylsuccinimides revealed antifungal properties. Interestingly, the challenging synthesis of enantiopure succinimides and N-substituted succinimides 3 featuring all-carbon quaternary centers (R1 ≠ H) was performed very recently by Tamura and collaborators using a chiral palladium- phenanthroline complex L5-PdCl2.[18] Reactions occurred in good yields, high enantioselectivities with a complete regioselectivity and the use of NH-free maleimides (R2 = H) is tolerated allowing for further post-functionalizations.

Feng and coworkers have reported a highly stereoselective approach to centrally and axially chiral succinimides relying on desymmetrization of N-(2-tert-butylphenyl)maleimide 2a via 1,4-addition of unprotected 3-substituted-2-oxindoles 4 under Lewis acid catalysis (Scheme 3).[19] The corresponding multichiral succinimides 5 featuring adjacent quaternary and tertiary stereogenic centers as well as a stereogenic C–N axis[20] are obtained in high yields and excellent enantio- and diastereoselectivities (up to 99% ee and > 19 : 1 dr). The origin of the observed stereoselectivity is rationalized by the formation of an organized transition state L6-TS in which all reaction partners and the chiral N,N’-dioxide ligand L6 are coordinated to the scandium(III) metal center. Such coordination enables the desymmetrization of 2a by enhancing the reactivity of β-position a and by favoring a preferential addition of 3-substituted-2-oxindoles on the Si-face of the maleimide substrate.

The transition metal-catalyzed enantioselective hydrogenation of 3-substituted maleimides 2 represents an interesting approach for the straightforward synthesis of relevant chiral succinimides 3. In the course of their development of novel bifunctional bisphosphine-thiourea ligands such as ZhaoPhos L7, the groups of Dong and Zhang reported the efficient rhodium-catalyzed enantioselective hydrogenation of various 3-aryl- and 3-methyl maleimides (R1 = Me, Ar) featuring N-substitution or not with enantioselectivities up to 99% ee (Scheme 4).[21] This methodology relies on the synergistic dual activation of both metal center and maleimide substrate through H-bonding and metal-olefin interactions as shown in L7-TS.

More recently, a dynamic kinetic resolution strategy relying on rhodium-catalyzed transfer hydrogenation was successfully exploited for the stereodivergent synthesis of chiral succinimides from 3-hydroxy-4-substituted maleimides 2b as challenging substrates (Scheme 5).[22] Interestingly, depending on the amount of triethylamine used to modify the existing keto-enol equilibrium, the corresponding syn- and anti-products 6 are selectively obtained with excellent yields and stereoselectivities using Noyori's type tethered rhodium catalyst (S,S)-L8-Rh(III) catalyst. Mechanistic studies revealed that the reaction proceeds through the C=O bond reduction rather than C=C bond with a hydride transfer occurring first followed by a proton transfer step. Final proton transfer from formic acid releases the syn-products 6a as a major compound and regenerates the active catalyst using catalytic amounts of Et3N (Scheme 5, right, R1 = aryl). With 3-hydroxy-4- aryl-N-substituted maleimides 2b (R1 = aryl), the corresponding anti-products 6b are then arising from epimerization of the syn-products 6a in the presence of a 5 : 2 azeotropic mixture of HCO2H/Et3N (Scheme 5, left). Under the same conditions, substrates presenting an alkyl substitution at the C4 position (R1 = alkyl) do not epimerize and furnish the corresponding syn-products 6a. Finally, reduction of both enol and imide moieties is observed using NH-free maleimides (R2 = H).

The Sharma's group has recently reported the oxidative palladium-catalyzed C–H alkenylation of chiral biaryl aldehydes 7 with N-substituted maleimides 2c affording the corresponding atropisomers 10 in good yields with 92%–99% enantiomeric excesses (Scheme 6).[23] L-tert-Leucine L9 is employed as chiral auxiliary directing group by forming a transient imine intermediate 8 triggering the subsequent carbopalladation to 9. As stereochemical outcomes of the reaction prevent classic β-H elimination, an acetate-mediated E1CB elimination type occurs releasing the targeted biaryl atropisomeric aldehydes 10, and regenerating the chiral directing group L9 and the palladium(0) easily oxidized in situ to palladium(II) by p-benzoquinone. While non-substituted biaryls at C6-position undergo a DKR pathway (yields > 50%), the reaction proceeds through a KR mechanism with moderate yields (yields < 50%) for sterically demanding substituted ones due to higher barriers to enantiomerization.

In the same vein, the stereoselective ortho-directed C–H alkylation of various benzamides 11 with prochiral N-arylmaleimides 2d was designed under rhodium catalysis by the group of Li in 2021 as an efficient way to install simultaneously both central and axial chirality in 12.[24] In a synergistic manner, the use of a chiral cyclopentadienyl rhodium catalyst (L10-Rh)2 favors an endo- coordination mode L10-TS of the olefin moiety during the stereodetermining migratory insertion step thus generating a chiral environment around the prochiral axis and subsequent remote control. Chiral induction proceeds under mild conditions with excellent enantio- and diastereoselectivities and a large substrate scope (Scheme 7).

Remote control of axial chirality is also observed in the course of the stereoselective transition metal-catalyzed hydrofunctionalization of prochiral N-arylmaleimide derivatives. In this field, Xu and coworkers recently described the challenging, chemo- and highly stereoselective palladium-catalyzed hydrosilylation of activated alkenes such as N-arylmaleimides 2e with diarylmethylsilanes 13 (Scheme 8).[25] The Si–C coupling occurs selectively over reductive O-hydrosilylation and reduction of the C=C bond through Chalk–Harrod mechanism to give silylated succinimides 14 featuring central and axial chirality. The high chemo-, enantio- and diastereoselectivities arise from specific aromatic interactions and steric repulsions with the substrate in L11-TS within the cavity-type structure of the chiral TADDOL-derived phosphoramidite ligand L11 in the course of the stereodetermining migratory insertion of the H–Pd–[Si] intermediate on the activated double bond.

2.1.2 Cycloadditions
Functionalized pyrrolidines and prolines are key frameworks for medicinal chemistry, valuable building blocks for natural product synthesis and as well efficient organocatalysts for stereoselective transformations. In this context, the stereoselective metal-catalyzed (3+2) cycloaddition offers a straightforward and atom-economical approach for the synthesis of these five-membered heterocycles.
Several research groups have reported the (3+2) cycloaddition of azomethine ylides with prochiral N-aryl substituted maleimides affording the desired fused pyrrolidines 15a,b with high enantio- and diastereoselectivities. Depending on reactions conditions, the N-substitution of the maleimide partner and above all, the nature of the ligand coordinated to the metal center, reactions yield various cycloadducts featuring four stereogenic adjacent centers and one remote C–N stereogenic axis. The catalytic activity of chiral sulfonamide phosphine ligands has been evaluated in silver-catalyzed (3+2) cycloadditions of various iminoesters with achiral N-arylmaleimides 2f. A high level of endo-selectivity was reported by Xu and collaborators using the multifunctional Xing-Phos ligand syn-(R, RS)-L12 based on a non-biaryl atropisomeric framework (Scheme 9, up).[26] As well, the easily available Ming-Phos ligand (S, RS)-L13 developed by Zhang and Liu groups is an efficient ligand for the atroposelective and endo-selective desymmetrization of prochiral N-(2-tert-butylphenyl)maleimide 2a (Scheme 9, down).[27] In line with these results, Wang and coworkers have described a similar silver-catalyzed atroposelective strategy to access such scaffolds using axially chiral TF-BiphamPhos ligand L14.[28] In the favored transition state L14-TS, the steric bulk of the PPh2 group of the chiral ligand guides the maleimide derivative to approach the Si face of the in situ generated azomethine ylide (C=N bond) forcing its congested tert-butyl group to be far away from the metal center. Additional stabilization through H-bonding interactions between the amino group of L14 and one of the carbonyl groups of the maleimide is also proposed.

Original and highly congested multichiral imidazolinyl[2.2]paracyclophanol N,O-ligands L15a,b have been designed by Guiry and coworkers for their application in stereodivergent zinc(II)-catalyzed (3+2) cycloaddition reactions of in situ generated azomethine ylides from iminoesters with achiral N-substituted maleimides 2c leading to fused pyrrolidines 15c,d (Scheme 10).[29] Based on relatively cheap Zn(II)-catalytic systems compared to silver ones, these reactions produce efficiently the corresponding cycloadducts in high enantio- and diastereoselectivities. The planar stereogenic element is at the origin of the high stereocontrol observed as the use of pseudo-enantiomeric ligands (S, S, SP)-L15a and (S, S, RP)-L15b which allows reaction to perform with similar results but with a reverse stereochemical outcome. Modification of the electronic properties of the ligand through variation of N-substituent and substitution of C-5 and C-4 positions of the imidazoline ring has an impact on both reaction efficiency and selectivity. The mechanism is proposed to undergo via transition states L15a,b-TS in which the zinc center is tetracoordinated to the N,O-ligand and azomethine ylide thus favoring an endo-approach of the dipolarophile. Depending on the nature of the ligand, the congested paracyclophane core and the phenyl group at the C-4 (L15a) or at the C-5 (L15b) positions force the latter to approach from respectively the Si-face or the Re-face of the ylide (C=C enolate bond).

Since their original report in 2022 on the first stereoselective synthesis of multichiral C–N atropisomers via a silver-catalyzed (3+2) cycloaddition reaction of activated isocyanides with prochiral N-arylmaleimides using a phosphine-squaramide ligand,[30] the groups of Liao, Qian and Bai have pursued this research topic in order to achieve a desymmetrizative diastereodivergent process to access selectively to four diastereomers starting from unique substrates by sole catalyst-control in a single step (Scheme 11).[31] More interestingly, the (3+2) cycloaddition reactions of prochiral N-quinazolinone maleimides 16 with isocyanoacetates 17 deliver novel N–N atropisomers 18a–d featuring three supplementary stereogenic centers in high yields and excellent stereoselectivities. While the use of their original bifunctional phosphine-squaramide ligand L16a is associated to a full endo-selectivity (Scheme 11, left), the opposite exo-cycloadducts are obtained by switching to the Trost's ligand L16b (Scheme 11, right). In a logical way, using the ent-L16a and ent-L16b results in the formation of the opposite enantiomers (Scheme 11, up). With L16a ligand, the favored transition state L16a-TS is stabilized by H-bonding interactions between the squaramide unit and the carbonyl groups of the maleimide moiety and a relatively small twist of the ligand (N–C–C–N dihedral angle in the cyclohexanediamine tether). The Trost's ligand L16b (Scheme 11, right) acts as a bidentate ligand and coordinates the silver center in a T-shape manner. In this case, the N–C–Ag bond in the favored L16b-TS is slightly distorted resulting in an optimized orbitals overlap and in a single stabilizing H-bond interaction. For more synthetic application, the reaction is compatible with the use of N-indole maleimide derivatives with the same efficiency.

The enantioselective synthesis of spiro-ϒ-lactams has attracted much attention as such frameworks exhibit remarkable properties at the origin of biological activity. Among the innovative methodologies available to achieve this goal, the development of an enantioselective C–H activation/annulation strategy with N-substituted maleimide partners is appealing since all the previous examples have been reported in racemic version.[8] In this context, Shi and coworkers have recently described an efficient and atom-economical reaction for the enantioselective synthesis of spiro-ϒ-lactams 20 relying on an original C–H alkenylation/ formal (4+1) spirocyclization sequence using N-quinolinyl benzamides 19 as directing groups and achiral N-substituted-maleimides 2c as electrophiles (Scheme 12).[32] Compared to classical and difficult to handle Co(III) complexes, a simple and commercially available Co(OAc)2 is employed under oxidative conditions for in situ generation of reactive Co(III) species. Chiral induction is realized by a bifunctional spiro-phosphoric acid (CPA, (S)-STRIP) L17 acting both as a ligand for the metal and as a H-bond donor with the carbonyl group of maleimide resulting in highly organized transition states L17-TS for enantiodiscrimination in the course of the reaction. A mechanism supported by deuteration experiments and kinetic isotope effect (KIE) studies is proposed to explain the exact role of this binary catalytic system. Firstly, ortho-directed C–H alkenylation of N-quinolinyl benzamide occurs to produce a first Co(III)-intermediate after subsequent oxidation. At this stage, the suitable chiral environment created by CPA L17 enables a first enantiodetermining step installing the spiro moiety through a syn-aminocobaltation or an aza-Michael addition. Tautomerization and final stereoselective CPA-assisted protonation of the resulting Co(III)-enolate yield the desired enantiopure spiro-ϒ-lactams. The methodology has been highlighted with the straightforward enantioselective synthesis of an aldose reductase inhibitor. More recently, in a context of sustainable chemistry, a related process has been designed by Ackermann and coworkers for the enantioselective synthesis of spiro-ϒ-lactams based on electro-oxidative cobalt catalysis in association with the readily available Binol phosphoric acid L18.[33]

2.2 Organocatalyzed functionalizations
2.2.1 Nucleophilic additions
The organocatalyzed enantioselective nucleophilic addition on maleimides 2c is quite popular and the most studied pro-nucleophiles are simple enolizable aldehydes 21 that can be activated as enamine intermediates using a chiral primary amine organocatalyst leading to functionalized succinimides 22 (Scheme 13). The group of Chinchilla and Nájera reported in 2012 the use of amine-guanidine C1 as efficient organocatalyst[34] while, in 2015, the same team employed amino- pyrimidine C2 to catalyze this reaction.[35] In these two cases, polar DMF-H2O solvent system was employed, preventing the activation of the maleimide substrate by the NH moiety of the organocatalyst via hydrogen-bonding. In complement to these previous results, Chinchilla, Gómez-Bengoa and Guillena designed a chiral amine-salicylamide C3 able to catalyze this reaction, in toluene as solvent, affording the desired enantioenriched product in good yields and generally high enantiomeric excesses.[36] In this case, theoretical calculations account for a preferred energetically favored transition state C3-TS where the catalyst activates both the electrophilic maleimide and the nucleophile via hydrogen-bond and covalent bond (enamine) interactions, respectively.

Impressive results for this reaction using isobutyraldehyde as the pro-nucleophilic partner were recently disclosed by Shim and co-workers who utilized a very low amount (0.01 mol%) of the amino-thiourea catalyst C4 in water, allowing to reach excellent yields and enantioselectivities for the four reported examples.[37] Related chiral 1,2-diamines C5 immobilized on a polystyrene support via a sulfonyl linker were successfully used in the present reaction by Szőllősi.[7] The supported catalysts were recyclable, keeping their activity up to four runs with constantly high enantioselectivities. The same team reported the use of a combination between simple aminoacids such as L-phenylalanine and inorganic materials such as bentonite or alumina allowing to reach high yields and excellent enantioselectivities.[38] This organic-inorganic hybrid material can also be prepared ex situ and recycled several times with small activity loss due to leaching of the organic catalyst in the reaction solvent.
Apart from catalyst derived from either trans-cyclohexan-1,2- diamine or 1,2-diphenylethan-1,2-diamine, other more complex organocatalysts have been used for the efficient realization of this transformation. This concept has been pioneered by Wennemers and collaborators in 2016[39] by designing the tripeptide C6 able to promote the Michael addition of simple α-unbranched aldehydes (R2 = H) to unprotected maleimide (R3 = H) with high stereoselectivities and yields.[40] Four years later, the group of Da described the use of tetrapeptide C9, which is able to deliver the desired adducts in high yields and enantioselectivities starting with N-substituted maleimides (R3 ≠ H).[41] α-Branched aldehydes can be employed even if the diastereoselectivity remained moderate in these cases. The mechanism is proposed to go via transition state C9-TS where the enamine intermediate adds on the Si face of the maleimide, which is activated via hydrogen-bonding by the terminal carboxylic acid function of C9. The intermediate issued from this transition state after carbon-carbon bond formation has been identified by high resolution mass spectrometry. The work of Zhao and colleagues in this field is important since not only aldehydes but also ketones could participate in this reaction. They demonstrated that the activity of a simple amino acid C7 could be greatly enhanced by forming MDOs (Modularly Designed Organocatalysts) with cinchona alkaloid thiourea C8.[42] In this example, the complementarity of the two organocatalysts is crucial with C7 forming the enamine intermediate with the aldehyde or ketone and C8 increasing the electrophilicity of the maleimide partner via hydrogen-bond interactions.
In complement to this wide variety of nucleophilic activations of simple carbonyl derivatives, Bencivenni's group was the first to exploit the powerful Michael acceptor reactivity of maleimides to design a unique remote control of axial chirality for accessing succinimides displaying both central and axial chirality.[43] Their method involves the aminocatalytic desymmetrization of prochiral N-arylmaleimides through the vinylogous Michael addition of 3-substituted cyclohexenones 23 as pronucleophiles to N-(2-tert- butylphenyl) maleimides 2g (Scheme 14).[44] The salt formed by aminoquinine derived catalyst C10 and N-Boc phenylglycine C11 catalyzes the enantioselective desymmetrization via dienamine activation, resulting in atropisomeric succinimides 24 with two adjacent stereocenters. Primary amine catalysis was crucial for achieving enantioselective desymmetrization with simultaneous and exclusive remote control of the stereogenic axis and the newly formed stereocenters, with moderate diastereoselectivity but consistently high enantioselectivity.

Based on these promising results, the same group developed complementary organocatalyzed desymmetrizations of N-(2-tert- butylphenyl) maleimides 2g to functionalized succinimides 27 or 28 through Michael addition of either 3-phenyloxindoles 25 (Scheme 15, up) catalyzed by C12[45] or 1,3-ketoesters 26 (Scheme 15, down) catalyzed by C13[46] allowing for the control of two adjacent stereogenic centers and one stereogenic C–N axis.

Recently, the team of Szőllősi described the enantioselective organocatalyzed nucleophilic addition of 1,3-dicarbonyls 29 on maleimides 2c (Scheme 16, up).[47] They used the primary amine-thiophosphoramide C14 as chiral bifunctional organocatalyst yielding the desired succinimides 30 bearing a tetrasubstituted center in good yields and high stereoselectivities. In particular, the diastereoselectivity is excellent in the case of cyclic β-ketoesters 31 leading to succinimides 32 (Scheme 16, down, > 99 : 1 dr). The reaction was next extended to acetyl acetone (not shown) and the corresponding succinimides were obtained, albeit slightly lower enantioselectivities were obtained with this pronucleophilic partner (83%—87% ee).

Isocyanoacetates 17 are very useful building blocks in organic synthesis as they can easily be transformed in nitrogen heterocycles or branched aminoacids.[48] These compounds were used as pronucleophilic partners towards maleimides 2c under hydrogen- bonding organocatalysis with catalyst C15 (Scheme 17).[49] Hence, chiral succinimides 33 bearing contiguous quaternary and tertiary stereogenic centers were produced efficiently. Hydrolysis under concentrated hydrochloric acid allows for the conversion of the isocyanide function into the corresponding primary amine, without erosion of the enantiopurity. A few years later, Bencivenni exploited his remote control of axial chirality via nucleophilic addition of isocyanoacetates 17a to prochiral maleimides 2g leading to multichiral succinimides 34 controlling two contiguous stereocenters and revealing the axial chirality.[46]

Finally, in a very recent contribution, Wang and collaborators exploited the organocatalyzed enantioselective Michael addition of pyrazolones 35 for the desymmetrization of prochiral N-pyrazolyl maleimides 2h (Scheme 18).[50] The optimal catalytic activity was found with chiral amino thiourea C16 in toluene at room temperature giving the expected bis-pentatomic C−N pro-atropisomeric N-pyrazolyl succinimidopyrazolones 36 featuring two stereogenic centers with good enantioselectivity but no diastereoselectivity when R3 = t-butyl. Surprisingly and without explanation, in the cases of R3 = i-propyl, phenyl or cyclohexyl the authors reported a > 20 : 1 dr. A subsequent bromination with N-bromosuccinimide (NBS) revealed the C−N stereogenic axis in 37 with conservation of the two stereogenic centers in moderate yield but with high diastereo- and enantioselectivity.

2.2.2 Cycloadditions
Cycloadditions are quite popular for the desymmetrization of maleimide substrates 2c acting as dipolarophiles.[51] Within this area, Morita-Baylis-Hillman (MBH) carbonates 38 can be used as C3 partners in stereoselective (3+2) cycloadditions, when activated by nucleophilic organocatalysis, chiral phosphines being privileged organocatalysts for this transformation. One pioneer example was reported by Lu and colleagues using chiral phosphine C17 which adds onto MBH carbonates with loss of CO2 and release of tert-butoxide anion, triggering the formation of an ylide intermediate after deprotonation (Scheme 19).[52] The latter undergoes formal (3+2) cycloaddition on maleimide dipolarophile which is activated by hydrogen-bonding as proposed in the transition state C17-TS to afford the desired functionalized bicyclic imides 39 in excellent yields and enantioselectivities. This reaction was also explored two years later by Zhong and collaborators who used chiral ferrocenylphosphine C18, albeit with less efficiency.[53] The same group greatly improved their results in designing related chiral ferrocenylphosphine C19, rendering the expected products in high yields and excellent enantioselectivities.[54] Interestingly, the opposite enantiomer was obtained using this catalyst, which was found to be air stable both in solution and in solid state.

Higher level of complexity was achieved by Wang and Lin who employed prostereogenic maleimides 2g for the (3+2) annulation process with MBH carbonates 38 (R1 = H) under nucleophilic organocatalysis with chiral phosphine C20 (Scheme 20).[55] The scope to fused bicyclic imides 40 is very broad (44 examples) and very high stereoselectivities have been obtained which can be explained by an almost exclusive approach of the ylide intermediate from the face opposite to the R2 aryl substituent. In addition, the authors used density functional theory (DFT) to estimate the barriers to diastereomerization for the fused adducts (R1 = R3 = H, R2 = tBu, EWG = CO2Me) and (R1 = H, R2 = Ph, R3 = o-Me, EWG = CO2Me) at 141.3 kJ·mol–1 and 135.8 kJ·mol–1, respectively. In this year, Wang and Yao reported the similar transformation with phosphine catalyst C21 using carbon disulfide (CS2) as solvent.[56] In their case they were able to introduce an extra substituent R1 = Ar in the MBH carbonate which led to the creation and control of an additional stereogenic carbon atom in the final fused bicyclic product, isolated as a single diastereomer, in high enantiopurity.

After their work using MBH carbonates, Lu's group developed a similar methodology using allenes 41 as the ylide precursor with maleimides 2c (Scheme 21).[57] They employed a closely related chiral phosphine C22 as catalyst which adds on the allene and affords the reactive ylide intermediate. As compared with MBH carbonates, there is no need of in situ base generation and the proposed transition state C22-TS accounts for the observed stereoselectivity. Interestingly, this method nicely complements the previous ones as regioisomeric bicyclic succinimides 42 (related to the position of the ester/ketone function) are produced efficiently.

Apart from C3 dipolarophile partners generated either from MBH carbonates or allenoates, Zhao's group proposed an enantioselective (3+2) cycloaddition of isatin-derived azomethine ylides with maleimides 2c (Scheme 22).[58] These isatin-derived azomethine ylides are generated by condensation between oxindoles 43 and primary amines 44, followed by bifunctional squaramide C23-catalyzed [1,2]-proton shift. Subsequently, ylide undergoes (3+2) cycloaddition under hydrogen-bonding activation with C23 which favors the Si face to approach the 3Si-4Re face of maleimides. In most cases, the chiral spirocyclic pyrrolidine-oxindoles 45 are produced in moderate yields, with excellent diastereo- and enantioselectivities.

Aside from (3+2) cycloadditions, the (4+2) cycloaddition also stands as a quite important way of desymmetrizing maleimide substrates. Bencivenni and co-workers exploited their concept of remote control of axial chirality via a formal Diels−Alder reaction in which the prochiral maleimide 2g acts as the dienophile towards the chiral dienamine in situ generated by condensation between acyclic enones 46 and primary aminoquinine catalyst C10 (Scheme 23).[59] The expected fused bicyclic succinimides 47 bearing a C−N stereogenic axis and three stereogenic centers are obtained with good yields and perfect control of the diastereo- and enantioselectivity. In favored transition state C10-TS, hydrogen-bond between the quinuclidinium ion and the carbonyl of the maleimide guides the exclusive diene approach opposite to the o-tbutylphenyl group accounting for the observed stereochemistry.

Inspired both by the previous work of Bencivenni and the report of Lee on the organocatalyzed synthesis of achiral phthalimides via Diels−Alder between enals and maleimides,[60] Mukherjee's team developed an atroposelective de novo arene synthesis when starting from prochiral maleimides 2g (Scheme 24).[61] The enals 48 first form the corresponding dienamine intermediate 49 by condensation with C24 and the subsequent stereo-determining (4+2) cycloaddition with maleimides gives the expected cycloadducts 50 featuring four stereocenters and a stereogenic C–N bond. Then, the base (DABCO) promotes the elimination of C24, giving the cyclic cyclohexa-1,3-dienes 51, which spontaneously aromatized under air to afford the final axially chiral phthalamides 52 in good yield and generally excellent enantioselectivity.

Instead of generating in situ a chiral dienamine, Hara and co-workers designed an alternative strategy based on the direct use of achiral dienamines 53, which undergo enantioselective Diels−Alder reaction with maleimides 2c catalyzed by a chiral pyridinium phosphamide C25•TfOH (Scheme 25).[62] This chiral Brønsted acid displays two close acidic protons that are essential to enhance catalytic activity and to control the stereoselectivity. These acidic hydrogen atoms are able to participate in a strong hydrogen-bonding network with the substrates for the (4+2)-cycloaddition which delivers the endo cycloadducts 54 in good yields and stereoselectivities. Interestingly, the addition of 0.5 equivalent of neutral C25 relative to C25•TfOH resulted in an increase of the enantioselectivity. This could be explained due to the pyridine moiety of C25 which inhibits racemization by neutralizing the achiral Brønsted acid formed in situ by protonation of the maleimide that could be capable to promote the racemic background reaction.

In complement, our group described a novel chiral NHC-catalyzed pseudo-three-component reaction between an enal 55 and two equivalents of a prochiral N-aryl maleimide 2h under oxidative conditions using either triazolium salts pre-C26 or pre-C27 as pre-catalysts (Scheme 26).[63] This reaction produces bis-succinimide adducts 56 with high stereocontrol, forming up to four consecutive stereogenic centers and two remote C–N stereogenic axes. The best results were achieved using an inorganic base to generate the reactive NHC precursor of the Breslow intermediate. This intermediate, upon in situ oxidation by 3,3'-5,5'-tetra-tert- butyl-4,4'-diphenoquinone (TTBDQ), formed the key chiral azolium dienolate. This triggered a formal (4+2) cycloaddition, followed by a highly diastereoselective Michael addition onto a second equivalent of N-aryl maleimide. The resulting products, which include multiple stereogenic elements and two challenging C–N axes in the pentatomic series, were obtained as a single diastereomer out of 64 possible stereoisomers with excellent enantioselectivity. In addition, upon treatment under basic conditions (tBuOK), the bis-adducts eliminate one equivalent of N-aryl maleimide affording the expected N-hydroxyaryl phthalimide atropisomers 57 with good yields and excellent retention of enantiopurity.

Another way to generate in situ reactive dienophile partners was reported by Zhao and Jiang with a C15- organocatalytic activation of 5H-oxazol-4-ones 58, which upon deprotonation forms the corresponding cyclic 2-azadienes, triggering the (4+2) cycloaddition with maleimides 2c to form a series of chiral oxa-bridged piperidone-fused succinimides 59 in good yields, perfect diastereoselectivities and good to excellent enantioselectivities (Scheme 27).[64] The presence of sodium phosphate salt is crucial, not only to act as a base for the deprotonation of the 5H-oxazol-4-ones, but also to play an important role in the favored transition state C15-TS by binding the catalyst and the heterodiene through hydrogen-bonding and multiple dipole-dipole interactions.

Previous examples involved either (3+2) or (4+2)-cycloadditions, but alternative combinations in particular (n+1) cycloadditions leading to spirocyclic compounds have been disclosed recently. In 2016, Rovis designed a domino photoisomerization/ intramolecular Stetter reaction exploiting the property of N-heterocyclic carbenes (NHC) to promote umpolung reactivity of aldehydes.[65] Hence, this formal (3+1) cycloaddition starts from functionalized maleimide bearing a (E)-configured enal function, the intramolecular Stetter reaction leading to the spiro-heterocycle 60 was optimized allowing for the identification of pre-catalyst pre-C28 as the best one in terms of yield and enantioselectivity (Scheme 28).[66] Photoisomerization using UV wavelength allowed to isomerize the enal, a prerequisite for this enantioselective cyclization. The reaction was carried out on a 2.5 g scale and was the key step for the total synthesis of three diastereomers of (–)-cephalimysin A.

Recently, this approach to enantioenriched spiro-succinimides 62 was also explored by Jindal, Mukherjee, and Biju, involving a formal (4+1)-cycloaddition. Hence, a highly efficient NHC-catalyzed Stetter-aldol domino sequence was developed with aromatic o-dialdehydes 61 and prochiral N-aryl maleimides, followed by an oxidation step (Scheme 29).[67] This two-step process produces atropisomeric N-aryl succinimides, which feature both a stereogenic C–N bond and a spirocyclic stereogenic center, achieving good yields and high enantiomeric excess values. The reaction mainly applies to o-tBu-N-aryl derivatives and uses 10 mol% of NHC, generated in situ by deprotonating the chiral triazolium salt pre-C27 with DIPEA. DFT calculations suggest that the enantio-determining step is the Michael addition of the transient Breslow intermediate on the electrophilic maleimide substrate.

3 Maleimide as Pronucleophilic Partners
3.1 From Morita-Baylis-Hillman reaction
As highlighted in previous sections, functionalizations of maleimides as electrophilic partners in additions or cycloadditions all lead to multichiral succinimide derivatives. However, given the great synthetic and biological relevance of the maleimide motif, its preservation after enantioselective functionalization constitutes a real synthetic challenge. To this endeavor, the main solution is to take advantage of the potential nucleophilic character of maleimides. Despite its high synthetic potential, this counterintuitive reactivity is largely underdeveloped, but has been the subject of significant developments in recent years, which will be detailed in this section.
The first work in this direction was published in 2013 by Chimni and collaborators,[68] who developed a highly efficient β-isocupreidine C29-catalyzed Morita−Baylis−Hillman reaction of maleimides 2c with diversely substituted N-protected isatins 63, providing 3-hydroxy-2-oxindole derivatives 64 in high yields and excellent enantioselectivity (Scheme 30, X = O).

On the basis of absolute configurations determined by X-ray diffraction analysis, the authors have proposed a mechanism to account for the observed enantioselectivity. Thus, the tertiary amine function of the catalyst adds to the maleimide to give first a chiral enolate intermediate, which undergoes an aldol-type process rendering the key betaine (Scheme 30). The enantioselectivity is governed by the stabilization through the establishment of hydrogen bonds between the oxyanion and the amidic carbonyl group of the isatin with the aromatic hydroxy group of the catalyst, in a spatial arrangement that limits steric interactions between the aromatic rings of the isatin and the catalyst. Two years later, they extended the same methodology to N-substituted isatin imine derivatives resulting in the first enantioselective organocatalyzed aza-Morita−Baylis−Hillman reaction involving maleimides as pronucleophiles. 3-Substituted-3-aminoindolin-2-ones were thus isolated in moderate to good yields and exhibiting high level of enantioselectivity (up to 99% ee) (Scheme 30, X = N-Boc).[69] Finally, Chen and colleagues adapted the methodology by slightly modifying the operating conditions to introduce 7-azaisatins (Scheme 30, X = O, Y = N) as partners, which proved to be better electrophiles than their isatin analogues.[70] The corresponding 3-hydroxy-7-aza-2-oxindoles were obtained in high yields and good to excellent enantioselectivities in only 24–48 h.
3.2 From α-aminomaleimides
More recently, it has been shown that another strategy to take advantage of the nucleophilic character of maleimides is to use their enamine derivatives. Based on this concept, Oriyama, Mori and colleagues have reported in 2021 the first enantioselective organocatalyzed Michael addition of α-aminomaleimides 2j to β-nitrostyrenes 65 (Scheme 31, right).[71] Thus, in the presence of only 1 mol% of a quinine-derived urea catalyst C30a, the corresponding chiral maleimides 66 are obtained in 48 h at 10 °C in very good yields and enantioselectivities (up to 92% ee). The study was supported by theoretical DFT calculations, which enabled to optimize the enantioselectivity of the reaction by modulating the nature of the substituent on the maleimide nitrogen atom, and to determine its mechanism. A transition state C30a-TS in which the nitroolefin is activated by the urea unit and the donor by the tertiary amine function of the catalyst rationalizes the formation of the major enantiomer with (R) configuration. In the year after this pioneer contribution, the group of Wang and Jin[72] identified Takemoto's catalyst C31 as an alternative of comparable efficiency and allowing for a substantial extension of the substrate scope.

In 2024, Oriyama's group adapted this organocatalyzed activation with C30b for the development of a Mannich reaction with N-Boc aryl imines 67 providing the corresponding adducts 68 in high yields and enantioselectivities (Scheme 31, left).[73] The method proved highly effective with arylamines, but seemed more limited in terms of yield with aliphatic imines, although enantioselectivity remains very high.
Unlike the synthesis of fused-bicyclic succinimides, methods for preparing bicyclic maleimides other than phthalimides are extremely rare and the use of α-aminomaleimides 2j in (3+3) annulations provides a highly effective solution to this synthetic problem. In this context, Hui and Qi and Duan's groups contemporaneously described efficient annulations of N-aryl-α-aminomaleimides as 1,3-C,N-bis-pronucleophiles with simple enals 69 in the presence of N-heterocyclic carbene catalysts (NHC) under oxidative conditions, for the synthesis of pyrrolo[3,4-b]pyridines 71 (Scheme 32).[74, 75] Remarkably, when an ortho-tbutyl aryl group is present on the nitrogen atom of the enamine function (R3 = tBu), highly atroposelective synthesis of C−N axially chiral fused heterocycles was possible (up to 99% ee), while other ortho-substituents (R3 = iPr, OMe, Br, I) proceeded with only low enantioselectivity (9% to 32% ee). The very high stereochemical stability of these products was confirmed both by experiments and DFT calculations, with barriers to enantiomerization estimated around 209 kJ/mol, which is higher than that of most C−N atropisomers. The proposed mechanism involves first the reaction of the enal partner with the NHC catalyst, in situ generated by deprotonation of the pre-ent-C27, affording an α,β-unsaturated acylazolium intermediate upon DQ-oxidation of the transient Breslow intermediate. Subsequent DBU promoted deprotonation of the N-aryl-α- aminomaleimide triggered a domino process involving 1,4-conjugate addition/proton transfert/tautomerization/lactamization to dihydropyridones 70 that occurs with release of the NHC catalyst. A final in situ DQ-oxidation gave access to a large series of the final enantioenriched C−N atropisomeric products 71. The only difference between the two protocols is that Qi and Duan's group uses additives such as molecular sieves and a thiourea to activate the α-aminomaleimide and facilitate its deprotonation by DBU.

Based on a similar (3+3) annulation strategy between racemic axially chiral α-aminomaleimides 2j and α,β-unsaturated acylazolium intermediates generated in situ from 2-bromoenals 72 by NHC-catalysis involving pre-ent-C27, Biju's group reported in 2022 a kinetic resolution (KR) with remote chirality control to furnish C–N axially chiral fused-dihydropyridinones 73 with up to 6/1 dr and > 98 % ee featuring a stereogenic center and leaving the enantioenriched starting material (P)-maleimides 2j with ee > 98% (Scheme 33).[76] The remote axial chirality control was governed both by the steric hindrance generated by the bulky ortho-tbutyl substituent of the aryl moiety of the substrate, and by the aminoindanol scaffold of the NHC catalyst as shown in the proposed transition state ent-C27-TS resulting in good to excellent enantioselectivities but moderate diastereoselectivities.

3.3 From α-hydroxymaleimides
Among maleimide derivatives used for their pronucleophilic character, α-hydroxymaleimides 2k proved to be highly functionalized scaffolds, capable of reacting with both an electrophile and a nucleophile. Wang's group was the first, in 2017, to take advantage of the synthetic potential of these substrates by developing an organocatalytic diastereo- and enantioselective (3+2) heteroannulation between α-hydroxymaleimides 2k and nitrosoarenes 74 for the highly stereoselective synthesis of chiral quaternary N-hydroxyindolines 75 (Scheme 34).[77] This unusual reactivity of nitrosoarenes, compared to their more conventional oxyamination or aminoxylation reactions, proceeds with excellent yields and enantioselectivities with the chiral bifunctional Rawal's type organocatalyst C23.

A few years later, Zhang and co-workers reported another illustration of the synthetic potential of α-hydroxymaleimides in (3+2) annulation strategies, by developing a highly enantioselective reaction with quinone monoimines 76 catalyzed by chiral phosphoric acid C32 (Scheme 35, right).[78] The corresponding succinimide fused dihydrobenzofurans 77, bearing two continuous quaternary stereocenters, were isolated in moderate to excellent yields, with moderate to high enantioselectivities, up to 99 % ee. Based on the absolute configuration of one of the products that was determined by X-ray crystal structure analysis, the proposed mechanism involved a domino process initiated by conjugated addition of the α-hydroxymaleimide followed by aromatization and a terminal intramolecular hemiacetalization. The control of the enantioselectivity is ensured in the first step, in which both reaction partners are simultaneously activated by the bifunctional chiral catalyst as in C32-TS. Contemporaneously, Wang's group described the same reaction sequence, but directly on quinones 78, using bifunctional amino thiourea organocatalyst C4 activating both the α-hydroxymaleimide by hydrogen-bonding and the quinone covalently, via the formation of an imine (Scheme 35, left).[79] Similarly high level of efficiency in terms of yield and stereoselectivity were observed for the formation of fused polyheterocycles 79 through the H-bonding network in C4-TS.

In the same year, Zhang's group extended this domino methodology to azonaphthalenes 80 as Michael partners to access succinimide fused indolines 81 in moderate to very good yields, with generally good enantioselectivities, that can reach 98 % ee in some cases (Scheme 36).[80] The reaction was conducted in presence of chiral squaramide-tertiary amine catalyst C33, activating the acceptor through an hydrogen-bonding transition state C33-TS while the tertiary amine promotes the enol reactivity of the α-hydroxymaleimides 2k.

Wang's group also demonstrated in 2020 that α-hydroxymaleimides 2k were good building blocks for the development of (4+2) cycloadditions, illustrated by the synthesis of chiral fused tricyclic hemiketals (Scheme 37). Thus, a series of polycycles containing chroman and succinimide frameworks were synthetized in high yields and high levels of diastereo- and enantioselectivities through cinchona alkaloid squaramide C23a-catalyzed reactions between α-hydroxymaleimides 2k and ortho-hydroxyphenyl para- quinone methides 82 to give chromanes 83 (Scheme 37, left).[81] Based on control experiments and the observed configuration of the products, the authors suggested a plausible hetero Diels− Alder reaction involving the in situ formation of an ortho-quinone methide intermediate (o-QM), activated by the catalyst through C23-TSa. Shortly after, the same group[82] also demonstrated the synthetic potential of α-hydroxymaleimides for the synthesis of chiral hemiketals with 1,2-oxazine skeleton 85, via inverse-electron-demand oxa-Diels−Alder reaction with nitrosoalkenes, in situ generated from α-bromoketoximes 84 by elimination of hydrobromic acid in basic conditions in the presence of C23a (Scheme 37, up). The desired bicyclic products 85 were obtained under mild conditions with excellent diastereoselectivities (up to 99 : 1 dr) and enantioselectivities (up to 99% ee).

In complement, Du and collaborators in 2022 proposed a highly efficient diastereo- and enantioselective synthesis of chiral chroman-fused pyrrolidinediones 87 using a similar formal (4+2) cycloaddition approach involving 2-hydroxynitrostyrenes 86 as electrophiles inspired by Wang's previous results from ortho-hydroxyphenyl para-quinone methides (Scheme 37, right).[83] In this case, the reaction catalyzed by a related squaramide catalyst C23b through a hydrogen-bonding network C23-TSb involving the two partners is likely to take place via a domino sequence initiated by a Michael addition, followed by a hemiacetalization step.
4 Conclusions and Perspectives
This update of the last decade highlights the still relevant importance of using ancestral maleimides as multi-functional synthetic platforms in modern stereoselective organic synthesis. Relying on recent developments of enantioselective catalysis, both the electrophilic character and the pronucleophilic behavior of easily available functionalized maleimides have been exploited for the elaboration of new enantioenriched multichiral heterocyclic architectures, featuring both central and C–N axial chirality, of high synthetic and biological potential. From enantioselective metal-catalyzed or organocatalyzed Michael additions to various annulations, simple electrophilic maleimides acting as C1 or C2 synthons can be converted to pentatomic- or hexatomic-fused heterocyclic derivatives by (3+2) or (4+2) cycloadditions as well as complex spirocyclic scaffolds by (4+1) annulations.
More recently, the counterintuitive pronucleophilic potential of maleimides has received a renewed interest thanks to organocatalyzed activations. In this field, although the Morita–Baylis–Hillman reaction constitutes a pioneer contribution for the desymmetrization of simple maleimides, α-hydroxy- and α-aminomaleimides largely open the way to the development of new transformations. Hence, simple enantioenriched disubstituted maleimides are accessible either by Michael or Mannich additions involving the C-pronucleophilic reactivity, while annulations with 1,3-bis-electrophiles render multichiral hexatomic-fused systems, in some cases incorporating central and C–N axial chirality, when α-aminomaleimides are concerned as 1,3-C–N pronucleophiles. Concerning α-hydroxymaleimides, their C-nucleophilic reactivity combined with their subsequent C=O electrophilicity make them peculiar 1,2-dipolarophiles allowing for (4+2) or (3+2) stereoselective annulations to fused multichiral dihydropyran or dihydrofuran derivatives, respectively.
For more than 150 years, the maleimide nucleus has continued to attract the curiosity of scientists from several fields triggering a wide range of applications in a wide cross section of disciplines. More particularly, from a synthetic point of view, the smart recent contributions concerning enantioselective functionalizations clearly show that the reactivity of maleimides is still up to date and will continue to fascinate researchers in the quest of new valuable enantiopure functionalized maleimide and succinimide derivatives opening new perspectives in synthesis, biology and material sciences.[84]
Acknowledgements
Financial support from Aix-Marseille Université, the Centre National de la Recherche Scientifique (CNRS), and Centrale Méditerranée is gratefully acknowledged.
Biographies

Muriel Amatore was born in Paris in 1979 and obtained her Ph.D. in 2006 with Dr. C. Gosmini (UPEC, France). She carried out three post-doctoral research stays in enantioselective organocatalysis (Princeton University, Prof. D.W. C. MacMillan) and organometallic catalysis (Ecole Polytechnique, Dr C. Gosmini/Université Pierre et Marie Curie, Dr. Corinne Aubert) before achieving a permanent position as assistant professor at UPMC (2010). In 2018, she moved to Aix-Marseille Université to join the group of Prof. Thierry Constantieux (iSm2, STeRéO). Her current research interests center on the development of transition metal-catalysed and organocatalysed processes for their application in stereoselective synthesis.
Damien Bonne was born in 1979 in Epinal, France, and obtained his PhD in 2006 from the ‘Institut de Chimie des Substance Naturelles’ (ICSN) under the direction of Prof. Jieping Zhu working on multicomponent reactions. After a postdoc in the group of Prof. Varinder Aggarwal (Bristol), he was appointed Lecturer at Aix-Marseille University in 2007. He was promoted Full Professor in 2022 and in 2023, he became “Junior Distinguished Member” of the French Chemical Society. His research focuses on the development of enantioselective methodologies for the control of axial and helical chiralities.
Thierry Constantieux was born in Pau, France, in 1968. After studying chemistry at the University Bordeaux I (France), he received a Ph.D. degree in 1994. He completed his Habilitation in 2004, at Aix-Marseille Université (France), where he is currently Professor, and Director of the ‘Institut des Sciences Moléculaires de Marseille’. His main research interest is focused on the development of new eco-compatible synthetic methodologies, especially organo-catalytic enantioselective domino and multi-component reactions from 1,3-dicarbonylcompounds, and their applications in heterocyclic chemistry.
Jean Rodriguez was born in Cieza (Spain) in 1958, and studied chemistry at the University of Aix-Marseille (France), he completed his Ph.D. in 1987, and his Habilitation in 1992. He is currently Professor and was appointed Director of the UMR-CNRS-7313-iSm2 until December 2023. His research interests include the development of multiple bond-forming transformations and their application in stereoselective organocatalysed synthesis. In 2021 he was awarded the “Grand Prix Emile Jungfleisch from the French Academy of Sciences”.

