

Functionalization of Boranes through Thiol/Oxygen Catalysis†
† Dedicated to the Special Issue of Boron Chemistry.
Comprehensive Summary
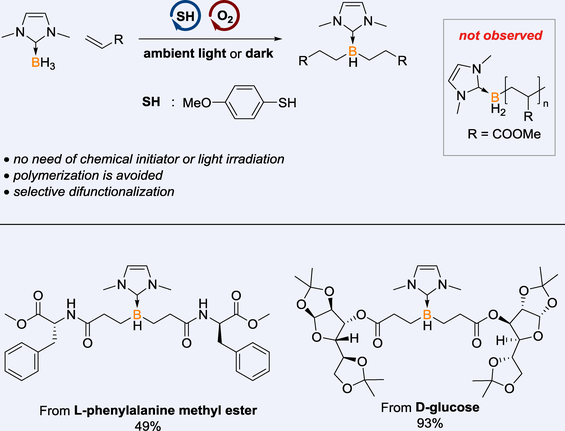
A greener and more convenient alternative to traditional methods for the generation of thiyl radical as hydrogen atom transfer (HAT) catalyst is developed, using molecular oxygen to oxidize thiol without the need for chemical initiators or light irradiation. The thiol/oxygen catalysis enables selective and efficient difunctionalization of borane.
Background and Originality Content
Thiyl radical has been recognized as a powerful hydrogen atom transfer (HAT) catalyst, which competently abstracts hydrogen atoms from carbon-hydrogen or element-hydrogen functionalities to deliver the corresponding carbon- or element-centered radical suitable for subsequent transformations.[1-3] Homolysis of disulfide represents a common method for generating the thiyl radical catalytic center.[4, 5] Starting with thiols, two prominent pathways to thiyl radicals involve single-electron oxidation by an excited photosensitizer, and deprotonation followed by photoinduced single electron transfer (Scheme 1a).[6-8]

However, these approaches require light irradiation, a proper photosensitizer, or specific substrates, thereby compromising overall efficiency and reaction scope. Therefore, exploring more convenient and milder methods to access thiyl radicals has drawn considerable research interest. Inspired by the seminal precedents of producing disulfides by oxidating thiols in air[9-13] and the formation of thiyl radicals from oxygen and thiols,[14-21] we hypothesize that a combination of thiol and molecular oxygen could afford desired thiyl radical for HAT catalysis (Scheme 1a). However, oxygen is often considered incompatible with radical reactions due to its propensity to terminate radical chains.[22, 23] To circumvent this limitation, careful modulation of the oxygen concentration may offer a viable solution.
Recent years have seen remarkable progress in organoboron chemistry, thanks to the broad application of organoboron compounds in synthetic chemistry,[24-33] boron-based pharmaceuticals[34-36] and optoelectronic materials.[37-40] As a complement to established transition metal catalysis, boron radical mediated borylation has emerged as a metal-free alternative for synthesizing a diverse array of organoboranes.[41-57] Despite these advancements, challenges persist. Firstly, when acrylates are employed as reactants, radical polymerization dominates as an undesirable side reaction.[58, 59] Secondly, the multi-functionalization of boranes is relatively underexplored, despite its potential to considerably enhance the complexity of bottom-up synthesized boranes.[60-70] Thirdly, the generation of boron radicals typically necessitates heating and/or light irradiation.[41-49] Taking the above aspects into consideration, we have initiated a study of borane difunctionalization with acrylates, employing thiol/O2 as the HAT catalysis system. Through elaborately screening, a green and mild protocol has been established with good compatibility. Excellent chemoselectivity is achieved, effectively overcoming the previously prevalent issue of radical polymerization (Scheme 1b).
Results and Discussion
In our proposed mechanism, oxygen initially oxidizes the thiol to generate a thiyl radical,[14-21] which then engages in HAT with borane to form a nucleophilic boron-centered radical. Nevertheless, the diradical nature of molecular oxygen makes it an effective radical trap, which may inhibit the further reaction of boryl radical with acrylates. Given the dual function of oxygen, our primary attempt is to determine the optimal amount of oxygen that balances its participation in thiol oxidation and its capacity as a radical trap. In the presence of 5.0 equiv. of air (around 1.0 equiv. of O2), 20 mol% 4-methoxythiophenol and diphenylamine as an additive, the reaction of NHC borane 1 with methyl acrylate at room temperature under ambient light for 2 h led to a mixture of mono- and di-alkylated borane (3a and 4a) in 19% and 4% 1H NMR yields, respectively. Reducing the loading of air significantly improved the efficiency and di-selectivity. When 0.5 equiv. of air (around 10 mol% of O2) was used, 4a was obtained in 88% 1H NMR yield. Further reducing the amount of air resulted in a relatively lower conversion (Scheme 2a). To further elucidate the role of thiol and molecular oxygen, control experiments were carried out (Scheme 2b). No desired product was detected in the absence of thiol, highlighting the critical role of thiol as the precursor for the thiyl radical.

Three possible pathways for thiyl radical generation are proposed: 1) the direct oxidation of thiol by oxygen to form thiyl radical,[14-21] 2) the reaction of borane with thiol generates a boryl sulphide intermediate, which undergoes B—S bond cleavage under photo-irradiation to afford thiyl radical and boryl radical simultaneously,[71-74] and 3) the oxidation of thiol to initially form a disulfide, followed by the subsequent homolysis of the disulfide under photo-irradiation, leads to the generation of thiyl radicals[9-13] (Scheme 2c, left). However, when boryl sulfide or disulfide was employed as a substitute for thiol, the desired alkylation products 3a or 4a were not detected, indicating that these compounds do not effectively serve as thiyl radical precursors under the reaction conditions (Scheme 2c, right).
In experiments conducted under ambient light but in an argon atmosphere, without additional air or oxygen, the yield of 4a decreased dramatically to 13%. In comparison, when the reaction was performed in dark but with the addition of 10 mol% O2, a yield of 65% for 4a was achieved. These observations highlight the essential role of a catalytic amount of oxygen, suggesting that photo-irradiation is not necessary for the reaction to proceed efficiently (Scheme 2b).
Considering the results from the above control experiments, it is evident that the primary pathway for thiyl radical generation in this borane dialkylation reaction is believed to be the direct oxidation of thiol by oxygen, supporting the predominance of the first proposed pathway. The thiyl radical trapping experiment was performed subsequently. In the absence of borane, stirring a mixture of thiol, oxygen, and methyl acrylate afforded the hydrothiolation product 5 in 13% yield. In contrast, without oxygen, there was no reaction observed. These results support the in-situ generation of thiyl radical from thiol and oxygen (Scheme 2d).
With the optimal conditions in hand (for detailed condition screening, see Supporting Information, Table S1), the substrate scope of this borane dialkylation reaction was explored (Table 1). With the thiol/O2 catalytic system, the reaction of borane with various acrylates affords dialkylated boranes 4a—4i in moderate to high yields. Sulfonyl groups were also tolerated, affording disubstituted boranes 4j and 4k. The generation of a mono-alkylated product 3k accounts for the low yield of 4k. 1,1-Disubstituted acrylates also worked well to provide 4l and 4m in the yields of 61% and 65%, respectively.

|
- a Reaction conditions A: reactions were conducted with NHC borane 1 (0.1 mmol), alkene 2 (0.36 mmol, 3.6 equiv.), Ph2NH (0.2 mmol, 2.0 equiv.) and 4-methoxythiophenol (0.02 mmol, 0.2 equiv.) in THF (2 mL) at room temperature under an argon atmosphere, with the addition of air (1.2 mL), and under ambient light for 2 h. b PhCOOH was used instead of Ph2NH. c The reaction time was 6 h. All yields are isolated yields after silica gel chromatography; the numbers in parentheses are 1H NMR yields before purification with 1,3,5-trimethoxybenzene as internal standard.
Besides acrylates, the scope of acrylamides was also examined. Boranes 4n and 4o were prepared in good yields and excellent di-selectivity. It was noteworthy that compound 4p bearing the primary amide proceeded equally well, demonstrating the method's compatibility with substrates containing active protons. On this basis, acrylamides derived from amino acids were reacted with borane 1 under the standard conditions, leading to the preparation of 4q and 4r in moderate yields. The relatively lower isolated yields can be attributed to the poor solubility of tetracoordinated borane derivatives of amino acids. This method holds potential for linking amino acids or peptides with organoboranes, which may find applications in biomedical chemistry.[8, 75] In addition, complex acrylate substrates derived from natural products, including fenchol, L-menthol, D-glucose, vitamin E, estrone, stigmasterol, 3β-hydroxyprogesterone and cholesterol, reacted well to afford 4s—4z in high yields.
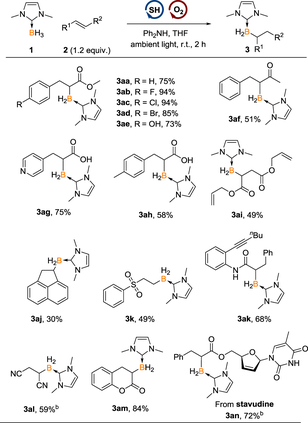
The reaction of borane 1 with more sterically hindered 1,2-disubstituted olefins, such as methyl cinnamate, produced mono- alkylated borane as the main product (Table 2). Under the optimal reaction conditions (for condition screening, see Supporting Information, Table S2), a series of mono-alkylated boranes were prepared selectively. Methyl cinnamate and derivatives bearing -F, -Cl, -Br and -OH groups were well compatible, offering 3aa—3ae in the yield > 73%. Besides, propenoic acids were also tolerated under the mild conditions to afford the desired borylation products 3ag and 3ah in good yields. In these reactions, C—B bonds were exclusively formed, with no O—B bond formation detected.[76]
|
- a Reaction conditions B: reactions were conducted with NHC borane 1 (0.1 mmol), alkene 2 (0.12 mmol, 1.2 equiv.), Ph2NH (0.2 mmol, 2.0 equiv.) and 4-methoxythiophenol (0.02 mmol, 0.2 equiv.) in THF (2 mL) at room temperature under an argon atmosphere, with the addition of air (1.2 mL) and under ambient light for 2 h. b PhCOOH was used instead of Ph2NH. All yields are isolated yields after silica gel chromatography.
For the olefin containing both electron-deficient and electron- rich double bonds, borylation occurred at the more electron-deficient internal C=C bond, indicating the involvement of nucleophilic boryl radical in the reaction pathway (3ai). Moreover, substrates with various functional groups such as nitrile (3al), alkynyl and amides (3ak) were compatible. Borylation of coumarin and stavudine derivatives also carried out smoothly to deliver 3am and 3an in 84% and 72% yields, respectively.
Detailed mechanistic study was performed to better understand the thiol/oxygen catalysed borane alkylation reaction (Scheme 3). The addition of 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) inhibited the formation of alkylated product 3a or 4a, with the detection of corresponding radical-trapping species by HRMS (Scheme 3a), indicating the involvement of thiyl, boryl, and carbon radicals in the reaction pathway. The latter originated from the attack of boryl radical at methyl acrylate. Moreover, the detection of compound 9 suggests the existence of a carbon radical generated from HAT of THF, which is supported by the deuterium labelling experiments (vide infra, Scheme 3e). A radical capture experiment was conducted using 1,1-diphenylethylene, which resulted in the formation of the anticipated mono-alkylation product (12) and trace amount of di-functionalized product (12’), suggesting the radical reaction nature (Scheme 3b).

Furthermore, the reaction was proven to go through a radical chain mechanism. Two parallel reactions were carried out (Scheme 3c). After reacting in dark for 10 min, one reaction was quenched, resulting in the detection of compound 4a in 43% yield. For the other reaction, oxygen within the reactor was removed through a freeze-pump-thaw cycle. The reaction then continued under argon and in the dark, leading to a further increase in the yield of 4a to 70%. This progression confirmed the self-propagating nature of the radical chain process. Moreover, to outline the hydrogen transfer diagram, deuterium labelling experiments were carried out. Parallel reactions using (NHC)BH3 1 and (NHC)BD3 1-D revealed a kinetic isotope effect (KIE) with a ratio of kH/kD of 4.0, indicating that HAT between thiyl radical and NHC borane is involved into the rate-determining step of the reaction (Scheme 3d). Under thiol/O2 catalysis, the reaction of 1-D with methyl acrylate in THF afforded the dialkylated product 4a-D in 36% yield and negligible deuterium incorporation at the C2 position. While in THF-d8, the deuteration level at the C2 position reached 46%, suggesting that the C2 carbon radical is capable of abstracting hydrogen/deuterium from the solvent THF. Furthermore, the addition of an excess (100 equiv.) of D2O under standard conditions resulted in the deuterated product 14 with 50% C2 deuteration rate (Scheme 3e).
Based on the above experimental results and the literature precedents, a plausible reaction mechanism is proposed in Scheme 3f. At room temperature, molecular oxygen oxidizes the thiol to form a thiyl radical. This thiyl radical then performs HAT from NHC-borane to generate a boryl radical. The generated boryl radical subsequently attacks the electron-deficient alkene, yielding a carbon radical intermediate. The carbon radical can then engage in HAT with the thiol to produce the alkylated product while simultaneously regenerating the thiyl radical catalyst.
The presence of proton sources, such as D2O, can induce proton exchange with thiol, resulting in the C2-deuterated product. The observed KIE value, which indicates a slower transfer rate for deuterium compared to hydrogen, aligns with the moderate rate of deuteration observed in the experiments. This suggests that the C2 carbon radical is capable of abstracting a proton from the solvent tetrahydrofuran (THF), leading to partial deuteration of the product. These observations are in accordance with the deuterium labelling experiments detailed in Scheme 3e. Finally, as the catalytic cycle continues, the regenerated thiyl radical can initiate a second alkylation, completing the catalytic circle for another round of reaction.
The thiol/O2 catalytic system was further employed in the sequential difunctionalization of borane, facilitating the efficient preparation of unsymmetrical dialkylated borane 15 in 45% yield (Scheme 4a). Additionally, the scalability of this methodology was showcased by the synthesis of compound 4a on a 1.3-gram scale, which proceeded with a high yield of 92%, analogous to that observed on a smaller scale (Scheme 4b). Most importantly, the thiol/oxygen catalytic system introduces a novel approach for the generation and utilization of thiyl radicals as hydrogen atom transfer (HAT) catalysts. Beyond the functionalization of boranes, the potential of this strategy will extend to other transformations, particularly those involving radicals of main group elements. This system's versatility is highlighted by the successful alkylation of silane, as depicted in Scheme 4c, marking an initial step toward broader applications.

Conclusions
In conclusion, we have achieved the efficient and selective difunctionalization of borane by employing a catalytic amount of thiol and oxygen. A thorough mechanistic investigation has revealed the pivotal role of oxygen in effectively producing the thiyl radical directly from the thiol. The potential of this thiol/oxygen catalysis extends beyond borane functionalization, as demonstrated by the successful functionalization of silane, suggesting wider applicability in various other chemical transformations.
Experimental
Under argon, a 10 mL reaction tube equipped with a magnetic stir bar and a septum was charged with NHC borane (11.1 mg, 0.10 mmol, 1.0 equiv.), alkene (0.36 mmol, 3.6 equiv.), Ph2NH (33.8 mg, 0.20 mmol, 2.0 equiv.) or PhCOOH (24.2 mg, 0.20 mmol, 2.0 equiv.) and THF (2.0 mL). 4-Methoxythiophenol (2.5 μL, 0.02 mmol, 0.2 equiv.) was then added. Air (1.2 mL) was injected into the system and the tube was sealed with Parafilm and stirred at room temperature for 2 h. After completion, the reaction was quenched with an excess amount of air, and the mixture was evaporated in vacuo. The resulting residue was purified by silica gel column chromatography to give dialkylated product.
Acknowledgement
This work was supported by research start-up fund (Project No. 4933621) from the Chinese University of Hong Kong. Mr. Weixuan Sun is thanked for checking the experimental procedure.


