

Nucleophilic Germylation of Stable π Bonds via Ge─Ge Bond Heterolysis
Graphical Abstract
A thermodynamically controlled nucleophilic germylation of stable π bonds was achieved by germyl anion generated in situ via heterolytic cleavage of the Ge─Ge bond in the presence of KOtBu. This methodology enables unprecedented multiple germylation of alkynes and internally selective germylation of alkenes, affording 1-alkyl-1-germylalkanes, 1,1-digermylalkanes, and 1,1,1-trigermylalkanes.

Abstract
Organogermanes have recently attracted a great deal of attention as building blocks for the synthesis of bioactive products, drugs, and functional materials. Metallo-germyls were classically synthesized and employed for nucleophilic germylation to form C─Ge bonds. However, their syntheses require highly reactive organometal reagents, and the scope of germylations involving metallo-germyls is limited due to competition with kinetically favored side reactions. Here, we present a regio/stereo-convergent nucleophilic germylation of stable π bonds by germyl anion generated in situ via heterolytic cleavage of the Ge─Ge bond of digermane (Ge─Ge) in the presence of KOtBu. This methodology affords unprecedented reactivity, enabling multiple germylation of alkynes and internally selective germylation of alkenes. These reactions are operationally simple, have broad functional group tolerance, and afford densely functionalized aliphatic organogermanes, such as 1-alkyl-1-germylalkanes, 1,1-digermylalkanes, and 1,1,1-trigermylalkanes, without any catalyst.
Introduction
The recent development of cross-coupling reactions and photocatalytic radical reactions of organogermanes has enabled the construction of sp3-rich molecules[1-5] useful as building blocks for the synthesis of bioactive natural products, drugs, as well as functional materials (Figure 1a).[6-11] In addition, owing to their unique 3D structure, good hydrophobicity, and low toxicity, aliphatic organogermanes have also attracted attention as potential pharmaceuticals.[12, 13] Nucleophilic germylation is a widely used method for the preparation of diverse organogermanes.[14] Historically, syntheses of metallo-germyls (R3Ge─M. M = Li, MgX, etc.)[15-20] from chlorogermane (R3Ge─Cl), digermane (R3Ge─GeR3), germylmercury (R3Ge─Hg), or germylhydride (R3Ge─H), followed by their reactions with electrophiles, have been utilized for C─Ge bond formation, with or without a catalyst (Figure 1b).[15-29] However, the synthetic reactions require an excess amount of highly reactive organometal reagents, and in addition, the high basicity of the synthesized metallo-germyls limits their reaction scope. As an alternative approach, in situ generation of germyl anions has been developed by combining readily available, storable, and easy-to-handle silylgermanes (Ge─Si) or digermanes (Ge─Ge) with fluoride anion (F−), driven by the formation of stable Si─F/Ge─F bonds. This methodology has largely replaced the use of metallo-germyls in nucleophilic germylation reactions, offering high functional group compatibility.[30-32] However, applicability for germylations of highly polarized (reactive) electrophiles, such as alkyl halides,[30] terminal enones,[30] and aryl onium salts,[31, 32] is limited (Figure 1c). Furthermore, simple applications of this protocol to compounds with stable π bonds, such as alkenes and alkynes, have proved unsuccessful due to insufficient activation of the Ge─Ge/Ge─Si bond by the F− anion.

Based on our research on interelement bond activations in Group 14 (Si─B and Sn─Si bonds),[33-35] we envisioned that an alternative activation method leading to higher Ge─Ge bond polarization would open up a new avenue for germylation of less activated π bonds. Preliminary density functional theory (DFT) calculations on hexamethyldigermane (Me3Ge)2 (Figure 1d) indicated that fluoride (F−) kinetically facilitates Ge─Ge bond activation but involves large endothermicity (>15 kcal mol−1). This is in good agreement with the experimental findings that germylation using digermanes (Ge─Ge) activated with fluoride anion is limited to highly polarized electrophiles. Among various nucleophiles, O-nucleophiles such as methoxide (MeO−) and tert-butoxide (tBuO−) were predicted to make the heterolysis kinetically and thermodynamically more favorable compared with fluoride, in accordance with the bond dissociation energies (BDEs)[36]: Ge─O 158 and Ge─F 116 kcal mol−1.
Thus, we speculated that the germyl anion species generated from digermanes via Ge─Ge bond activation by O-nucleophiles would expand the accessible chemical space of organogermanes. Indeed, our model calculations suggested that nucleophilic germylations of both phenyl alkene (C═C) and alkyne (C≡C) via alkoxide-mediated Ge─Ge bond cleavage would proceed exothermically (ΔG < 0 kcal mol−1), whereas the use of fluoride anion leads to endergonic reactions (ΔG > 0 kcal mol−1). However, despite the potential synthetic advantages, Ge─Ge bond activation of digermane with an alkoxide anion has so far only been used in germyl metal synthesis[19] and has not been employed in germylation reactions. Herein, we report in situ generation of germyl anion from digermanes (Ge─Ge) via Ge─Ge bond cleavage with tert-butoxide (KOtBu). Further, we show this enables unprecedented germylations with high functional group compatibility: internal-selective hydrogermylation of aryl alkenes and multiple germylation of aryl alkynes, expanding the accessible chemical diversity of aliphatic organogermanes (Figure 1e).
Results and Discussion
We first attempted nucleophilic germylation of styrene 1aa, which is theoretically predicted to proceed exothermically and might compensate for the energy loss incurred in the Ge─Ge bond heterolysis with O-nucleophiles, by using hexamethyldigermane (2) in the presence of a stoichiometric amount of base (Table 1). Fluoride sources (TBAF and CsF), which were previously used to generate germyl anion species,[30-32] resulted in no reaction, with recovery of 1aa (entry 1). In contrast, hydrogermylation of 1aa proceeded in the presence of KOMe or KOtBu, affording 1-methyl-1-germyl alkane 3a in 56% and 69% yields, respectively, with complete regioselectivity, though sodium alkoxides (NaOMe and NaOtBu) hardly promoted germylation of 1aa (entries 2–4). The yield of 3a was improved to 86% by the addition of 18-crown-6, which captures the potassium cation to generate counter-cation-free germyl anion (entry 5). These results demonstrated that the present equilibrium system combining Ge─Ge and KOtBu could achieve nucleophilic germylation of less activated π bonds, which is conventionally challenging without the use of a catalyst.[37-48]
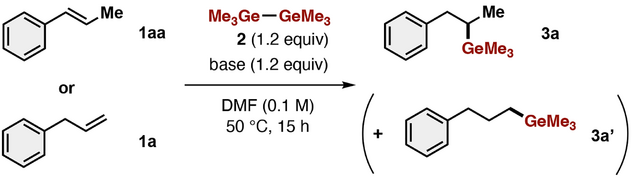 |
||||
| Entry | Alkene | Base | 3a Yield (%) | 3a’ Yield (%) |
|---|---|---|---|---|
| 1 | 1aa | CsF or TBAF | 0 | – |
| 2 | 1aa | NaOMe or NaOtBu | Trace | – |
| 3 | 1aa | KOMe | 56 | – |
| 4 | 1aa | KOtBu | 69 | – |
| 5 | 1aa | KOtBu + CE | 86 | – |
| 6 | 1a | CsF or TBAF | 0 | 0 |
| 7 | 1a | NaOMe or NaOtBu | Trace | 0 |
| 8 | 1a | KOMe | 56 | 0 |
| 9 | 1a | KOtBu | 67 | 0 |
| 10 | 1a | KOtBu + CE | 81 | 0 |
| 11 | 1a | KOtBu + ethylene glycol | 0 | 0 |
| 12 | 1a | KOtBu + triglyme | 70 | 0 |
| 13 | 1a | KOtBu + tetraglyme | 65 | 0 |
| 14 | 1a | KOtBu/blue LEDsb) | 79 | 0 |
| 15 | 1a | KOtBu/HMPAc) | 84 | 0 |
- a) 0.10 mmol scale reactions. Yields were determined by 1H-NMR analysis.
- b) Irradiation with blue LEDs instead of heating at 50 °C.
- c) HMPA at room temperature instead of DMF at 50 °C. TBAF = tetrabutylammonium fluoride; CE = 18-crown-6; HMPA = hexamethylphosphoric triamide.
During further investigations of the unique reactivity/selectivity of Ge─Ge/tBuO−, we discovered an internal-selective germylation of phenyl alkenes. Although hydrogermylation of alkenes is one of the most straightforward methods to obtain various aliphatic organogermanes, the reaction is limited, being linear-selective.[37-49] The present equilibrium system of Ge─Ge/KOtBu can achieve thermodynamically controlled germylation, in contrast to metallo-germyls (Ge─M), where the reaction is kinetically controlled. We found that KOMe or KOtBu dramatically promoted hydrogermylation of allylbenzene (1a) with complete internal selectivity to afford 3a (Table 1, entries 8 and 9) by base-mediated isomerization of terminal alkene to internal alkene[50] and in situ-generated KGeMe3-mediated germylation, while fluoride sources (CsF and TBAF) and sodium alkoxides (NaOMe and NaOtBu) were ineffective (entries 6 and 7). Addition of 18-crown-6 increased the yield of 3a (entry 10) to 81%, representing the optimal condition. Screening of polyethers revealed that 18-crown-6 is the best additive among those tested (entries 11–13). In line with our previous report on photoexcited stannyl anion species,[34] we found that conducting the present germylation reaction under irradiation with blue LEDs further increased the yield of 3a to 79% in the absence of 18-crown-6 (entry 14). Examination of several solvents revealed that HMPA, which can capture the potassium cation like 18-crown-6, also accelerated the present germylation, affording 3a in 84% yield (entry 15).
Representative results illustrating the scope of the present internal-selective hydrogermylation are summarized in Figure 2a. Compounds bearing a variety of substituents (o-/m-/p-tolyl, p-anisyl, and naphthyl groups) were converted to the corresponding 1-methyl-1-germyl alkanes 3a–3g in good yields without formation of undesired byproducts. This germylation could be performed on a preparative scale (1 mmol). Internal alkene 1h can also participate in this germylation to afford organogermane 3h bearing tertiary carbons. In contrast, octene 1i, which possesses no aryl rings, was not available, probably due to the difficulty of isomerization from terminal to internal alkene. Not only allylbenzene derivatives, but also but-3-en-1-ylbenzene (1k) and pent-4-en-1-ylbenzene (1l), having longer linkers between the phenyl and alkene groups, were also available, affording 1-phenyl-2-germyl alkanes (3k and 3l) with complete chemo- and regioselectivity. The position of the C═C bond in the starting phenyl alkenes had no influence on the products, i.e., 1k’ and 1l’ afforded the same products, 3k and 3l, respectively, even when a mixture of 1k/1k’ or 1l/1l’ was used as starting materials. 1,4-Diallylbenzene (1j), 1-allyl-4-(but-3-en-1-yl)benzene (1m), and 1,4-di(but-3-en-1-yl)benzene (1n) underwent double germylation at the internal positions, providing digermylated products 3j, 3m, and 3n.

This method also enabled deuteriogermylation of alkenes by simply changing the solvent from DMF to DMF-d7, affording 3a-D and 3k-D from allylbenzene (1a) and 3-en-1-ylbenzene (1k), respectively, with high deuteration ratios (up to 94%) (Figure 2b). In contrast, quenching with D2O did not provide any deuteration products. These results suggested that the present germylation proceeds via addition of in situ-generated germyl anions and protonation from DMF. In addition, the internal-selective hydrogermylations can be combined with photocatalytic radical-mediated transformations,[1-5] constructing sp3-rich chemical molecules from C─Ge bonds. Various terminal alkenes bearing a phenyl group (1a, 1k, and 1l) were transformed into the internally alkylated compounds (5aa, 5ka, and 5la) with alkene 4 with complete regioselectivity (Figure 2c).
As the germyl anion prepared from digermanes via Ge─Ge bond cleavage with tBuO− proved to be an excellent germylation reagent for phenyl alkene derivatives, we next examined whether its scope could be extended to phenylacetylene derivatives. Pleasingly, we found unique reactivity/selectivity in the terminal-selective multigermylation of a terminal alkyne [1-butyl-4-ethynylbenzene (6a)] (Table 2). Although many germylation reactions of alkynes with or without the use of a catalyst have been reported,[51-64] this is the first example of digermylation/trigermylation of terminal alkynes.
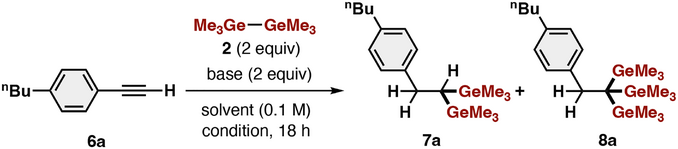 |
|||||
| Entry | Base | Solvent | Conditions | 7a Yield (%) | 8a Yield (%) |
| 1 | TBAF or CsF | DMF | rt | 0 | 0 |
| 2 | NaOMe or KOMe | DMF | rt | 0 | 0 |
| 3 | KOtBu | DMF | rt | 13 | 21 |
| 4 | KOtBu + CE | DMF | rt | 82 | 0 |
| 5 | KOtBu + CE | DMF | 50 °C | 81 | 0 |
| 6 | KOtBu + CE | DMF | Blue LEDs | 91 | 0 |
| 7 | KOtBu | HMPA | 50 °C | 79 | 0 |
| 8b) | KOtBu | HMPA | 50 °C | 91 | 0 |
| 9c) | KOtBu + CE | DMF | Blue LEDs | No reaction | |
- a) 0.10 mmol scale reactions. Yields were determined by 1H-NMR analysis.
- b) 0.5 equiv of MeOH was added.
- c) Me3Si─SiMe3 or Me3Si─SnnBu3 was employed instead of 2.
Fluoride sources (TBAF and CsF) and methoxide sources (NaOMe and KOMe) did not promote any germylation (entries 1 and 2). In contrast, multiple hydrogermylation of 6a proceeded in the presence of KOtBu, affording previously elusive 1,1-digermylalkane 7a and 1,1,1-trigermylalkane 8a in 13% and 21% yields, respectively (entry 3). The yield of 7a was drastically improved to 82% with complete regioselectivity by the addition of 18-crown-6 (entry 4). Conducting the present germylation reaction under irradiation with blue LEDs further increased the yield of 7a to 91%, whereas heating was not effective (entries 5 and 6). Examination of several solvents revealed that HMPA, which can capture the potassium cation also accelerated the present germylation, affording 7a in 91% yield in the presence of 0.5 equiv of MeOH and 79% yield in the absence of MeOH (entries 7 and 8). Interestingly, other Group 14 element─element bonds, such as Si─Si and Si─Sn did not promote any silylation or stannylation (entry 9), indicating that the equilibrium system of Ge─Ge/KOtBu is crucial for this unique reactivity.
The substrate scope of these multiple germylations is illustrated in Figure 3a,b. For dihydro-digermylation of terminal alkynes, we used a photoinduced condition (condition A) and a thermally induced condition (condition B), depending on starting materials. Various phenylacetylenes with an electron-donating group (p-nBu, o-/m-/p-Me, p-tBu, p-alkoxy, p-amino, and p-alkyl), as well as an electron-withdrawing group (p-aryl), were compatible with the reaction, affording the desired products 7a–7n in good to excellent yields. Naphthalene (6o) and heteroaromatics such as pyridine (6p and 6q) and thiophene (6r and 6s) were also applicable. 6t bearing two acetylene moieties could be subjected to double digermylation reactions to afford quadruply germylated alkane 7t. In contrast to aryl alkynes 6a–6t, aliphatic alkyne 6u was unreactive. This dihydro-digermylation could also be conducted on a preparative scale (1 mmol), giving over 250 mg of 7f.

In addition to terminal alkynes, diaryl internal alkynes 6v–6x provided trans-1,2-digermylated alkenes 7v–7x in good to high yields with complete stereoselectivity using the combination of Ge─Ge/KOtBu (condition C). Moreover, the dihydro-trigermylation of terminal alkynes proceeded with 1 equiv of KOtBu under photoinduced or thermally induced conditions (conditions D and E), giving the corresponding 1,1,1-trigermylalkanes 8a, 8j, 8o, and 8p in high yields, accompanied with small amounts of 1,1-digermylalkanes 7. Furthermore, 9a–9c bearing both acetylene and alkene groups underwent terminal-selective dihydro-digermylation of alkynes and internal-selective hydrogermylation of alkenes, producing highly germylated aliphatic compounds 10a–10c in good yields (Figure 3c). Additionally, the digermylated 7f was transformed into gem-chloro-germylated alkane 12f by photocatalytic monochlorination, affording a derivative of high-value modular C(sp3) platforms (Figure 3d).[4] Notably, all the products of 1,1-digermylalkanes 7/10 and 1,1,1-trigermylalkanes 8 were obtained with complete regio-/stereoselectivity with the present methodology.
Experimental mechanistic studies of the present dihydro-digermylation using the Ge─Ge/tBuO− system were conducted. Time course experiments using 6a revealed that the 1,1,1-trigermylalkane 8a is produced in the early stage and is converted into 1,1-digermylalkane 7a in the late stage (Figure 4a). The use of NaOtBu instead of KOtBu and 18-crown-6 did not give either 7a or 8a but afforded terminally germylated alkyne 6a–Ge in 90% yield (Figure 4b). Both 1,1,1-trigermylalkane 8a and terminally germylated alkyne 6a–Ge furnished 1,1-digermylalkane 7a under the standard conditions (Figure 4c), indicating that they are intermediates in the present dihydro-digermylation. Deuterium labeling experiments using DMF-d7 showed that the hydrogens on the benzyl position were exchanged to deuteriums from both 6e and 7e, while the hydrogen on the digermylated carbon was deuterated only from 6e (Figure 4d). These results indicate that the terminal proton in 1,1-digermylalkane 7 should be derived from the solvent, and 7 is in equilibrium with intermediates deprotonated at the benzyl position under the reaction conditions that we employed.

A plausible mechanism is illustrated in Figure 4e. As shown in Figure 1d, the combination of Ge─Ge/tBuO− makes the germyl anion (Me3Ge−) kinetically accessible under these reaction conditions. First, terminal alkyne 6 is subjected to monogermylation through acetylide I, giving intermediate II (6-Ge). Second germylation of II with Me3Ge− affords a vinyl anion intermediate III, which is protonated by tBuOH (derived from the terminal proton of 6) to give 1,1-digermylalkene IV. These processes (II → III → IV) would provide a compensating energy gain for the generation of Me3Ge− from Ge─Ge/tBuO−. Then, the third germylation of IV with Me3Ge− affords a benzyl anion intermediate V, which is successively protonated with DMF to furnish 1,1,1-trigermylalkene 8, providing a compensating energy gain for the generation of Me3Ge−. Finally, 8 gradually undergoes protodegermylation with DMF and tBuO− via intermediate VI to provide 1,1-digermylalkane 7, which is in equilibrium with deprotonated intermediate VII in DMF.
The rate-determining step is believed to be the formation of IV from II (II → III → IV), because (i) even the use of a weaker base (NaOtBu), not affording 8, could give II (Figure 4b), (ii) no germylated alkenes, including IV, were detected during the reaction, and (iii) the vinyl anion III should be the most unstable intermediate in the proposed mechanism, and the photo or thermal energy used in the germylations would be necessary to accelerate this step. Overall, the present multigermylation reaction should be thermodynamically controlled.
Finally, we performed DFT calculations, combined with the global reaction route mapping (GRRM) program,[65] to systematically investigate the possible reaction pathways for the present multigermylation at the M06/6-31+G* level of theory, using model compounds (phenylacetylene 6e, digermane 2, and KOtBu in DMF) (Figure 5a,b). The initial process for in situ generation of germyl anion (IM2) from Ge─Ge/KOtBu (IM1) in DMF would proceed kinetically at room temperature, albeit with an energy loss of 10.5 kcal mol−1. Indeed, in situ NMR measurements of the mixture of 2, KOtBu, and 18-crown-6 failed to detect methyl signals corresponding to KGeMe3 or tBuOGeMe3, in good agreement with our DFT calculation indicating that this equilibrium is thermodynamically unfavored (Figure S1). The starting alkyne 6e undergoes deprotonation and the first germylation at the terminal position with the release of tert-butanol (from IM3 to IM5). The assumption of monomeric tert-butanol, though it is at least dimeric in reality, would result in overestimation of the destabilization energy, but in any event, the energy loss of 10.5 kcal mol−1 in the heterolysis of the Ge─Ge bond and the total energy loss of 12.3 kcal mol−1 from IM1 are compensated by a significant energy release upon addition of the germyl anion to the C≡C bond of IM5 to afford the digermylated intermediate IM6. This second germylation requires the highest activation energy (26.0 kcal mol−1 from IM1) in the overall reaction process but is irreversible (ΔG‡rev = ca. 30 kcal mol−1), and this determines the regioselectivity. The vinyl anion IM7 is smoothly protonated by tBuOH to afford more stable IM8. Finally, the third germylation of the remaining C═C bond of IM9 is irreversible with a small activation energy of 3.1 kcal mol−1, giving the desired trigermylated intermediate IM10. The rate-determining step is thus the second addition of in situ-generated germyl anion, where TS5_6 required the highest activation energy, in accordance with the experimental results. Overall, the present multigermylation reaction is thermodynamically controlled with a large exothermicity of −43.8 kcal mol−1, in contrast to kinetic control in the case of metallo-germyls.

Conclusion
In conclusion, we have achieved in situ generation of germyl anion species via Ge─Ge bond heterolysis by alkoxide. The present protocol allows unprecedented germylations of stable π bonds, such as internal-selective hydrogermylation of aryl alkenes and terminal-selective multiple germylation of aryl alkynes, opening up new access to synthetically challenging 1-alkyl-1-germylalkanes, 1,1-digermylalkanes, 1,1,1-trigermylalkanes, and multiply-germylated compounds. In contrast to conventionally employed metallo-germyls, which provide kinetically controlled nucleophilic germylations, this combination of Ge─Ge/tBuO− achieves thermodynamically controlled nucleophilic germylations, providing a complementary tool for the preparation of a wide range of organogermane compounds. These mechanistic insights are confirmed by detailed experimental and theoretical studies. We believe that this work not only provides methodology for nucleophilic germylation reactions of stable π bonds but also showcases the value of theoretical investigations in providing a basis for designing and establishing a variety of nucleophilic reactions. Further studies to expand the reaction scope and to apply this approach for the synthesis of a range of functional molecules, including sp3-rich bioactive compounds, are in progress.
Acknowledgements
This work was partly supported by the JSPS Grants-in-Aid for Scientific Research for Transformative Research Areas (22H05125 to M.U.), the JSPS Grants-in-Aid for Scientific Research (A) (22H00320 to M.U.), the Transformative Research Areas (24H01839 to Y.N.), the Transformative Research Areas (24H01067 to Y.N.), the JSPS Grants-in-Aid for Scientific Research (B) (23H01956 to Y.N.), the JSPS Research Fellowships for Young Scientists (22J22969 to K.S.), and the JST FOREST (JPMJFR221Y to Y.N.). This work was also partly supported by grants from the Asahi Glass Foundation, the NAGASE Science Technology Foundation, the Naito Foundation, the Chugai Foundation, and the Uehara Memorial Foundation (to M.U.), the Asahi Glass Foundation, and the Uehara Memorial Foundation (to Y.N.).
Conflict of Interests
The authors declare no conflict of interest.
Open Research
Data Availability Statement
The data that support the findings of this study are available in the Supporting Information of this article.

