

2D Metal/Graphene and 2D Metal/Graphene/Metal Systems for Electrocatalytic Conversion of CO2 to Formic Acid
Graphical Abstract
Strong covalent bonding between metal monolayer and graphene driven by the sp and d orbital hybridization, 2D metal/graphene (M/G) system is investigated. The charge transfer from metal to graphene allows for the electrodeposition of another metal film, thus forming metal/graphene/metal (M/G/M) system. These 2D hybrid systems exhibit excellent activity and selectivity toward formic acid production over competitive hydrogen evolution reaction.

Abstract
Efficiently transforming CO2 into renewable energy sources is crucial for decarbonization efforts. Formic acid (HCOOH) holds great promise as a hydrogen storage compound due to its high hydrogen density, non-toxicity, and stability under ambient conditions. However, the electrochemical reduction of CO2 (CO2RR) on conventional carbon black-supported metal catalysts faces challenges such as low stability through dissolution and agglomeration, as well as suffering from high overpotentials and the necessity to overcome the competitive hydrogen evolution reaction (HER). In this study, we modify the physical/chemical properties of metal surfaces by depositing metal monolayers on graphene (M/G) to create highly active and stable electrocatalysts. Strong covalent bonding between graphene and metal is induced by the hybridization of sp and d orbitals, especially the sharp  ,
,  , and
, and  orbitals of metals near the Fermi level, playing a decisive role. Moreover, charge polarization on graphene in M/G enables the deposition of another thin metallic film, forming metal/graphene/metal (M/G/M) structures. Finally, evaluating overpotentials required for CO2 reduction to HCOOH, CO, and HER, we find that Pd/G, Pt/G/Ag, and Pt/G/Au exhibit excellent activity and selectivity toward HCOOH production. Our novel 2D hybrid catalyst design methodology may offer insights into enhanced electrochemical reactions through the electronic mixing of metal and other p-block elements.
orbitals of metals near the Fermi level, playing a decisive role. Moreover, charge polarization on graphene in M/G enables the deposition of another thin metallic film, forming metal/graphene/metal (M/G/M) structures. Finally, evaluating overpotentials required for CO2 reduction to HCOOH, CO, and HER, we find that Pd/G, Pt/G/Ag, and Pt/G/Au exhibit excellent activity and selectivity toward HCOOH production. Our novel 2D hybrid catalyst design methodology may offer insights into enhanced electrochemical reactions through the electronic mixing of metal and other p-block elements.
1 Introduction
The world's heavy dependence on fossil fuels has resulted in a very significant increase in atmospheric CO2, a major contributor to global climate change. Therefore, converting CO2 into value-added hydrocarbon fuels is an important strategy for mitigating the greenhouse effect and is expected to play a key role in the hydrogen-based energy society.1 Among the potential products of electrochemical CO2 reduction, formic acid (HCOOH) stands out as a promising option. HCOOH has a high hydrogen density of 53 g H2 per liter, surpassing the milestone set by Department of Energy, and is a viable liquid storage and delivery option for hydrogen in fuel cell applications.2 Furthermore, HCOOH is non-toxic, environmentally friendly, and has low flammability, making it an ideal energy carrier that can be transported easily at ambient conditions. These favorable properties make HCOOH a strong candidate for hydrogen storage technology.2a, 3
However, the selectivity challenge arising from the competitive hydrogen evolution reaction (HER)4 and the high overpotential required on conventional noble metal-based catalysts, such as Pt, Pd, Rh, and Ag,5 hinder the commercialization of CO2RR. Since either too strong or too weak binding energy of the formate (HCOO) intermediate specie leads to poor electrocatalytic activity,6 achieving a moderate binding energy of the HCOO intermediate species is essential for efficient electrocatalytic activity.
In this context, modifying the electronic structure of the active site can be a promising strategy to attain such moderate binding energy. Various electrocatalysts, including metal alloys,6a, 7 metal oxides,8 and perovskite oxides9 have been reported to promote CO2RR, but there remains significant improvement needed for commercial viability. For example,  nanoalloy catalysts reported by Koper et al. for CO2 and HCOOH conversion suffer from CO poisoning.10 The
nanoalloy catalysts reported by Koper et al. for CO2 and HCOOH conversion suffer from CO poisoning.10 The interface has been identified as an active site for CO2 activation and CO2RR.8a These catalysts utilize lattice mismatch and interatomic mixing between two or more different elements to alter the electronic structure of the active site, promoting the catalytic reaction. However, they typically require high noble metal loadings, whereas reducing the noble metal loading of the catalyst is essential to reduce costs. Efforts towards developing cost-effective, highly active, and selective catalysts for CO2RR are critical for the widespread adoption of this promising technology.
interface has been identified as an active site for CO2 activation and CO2RR.8a These catalysts utilize lattice mismatch and interatomic mixing between two or more different elements to alter the electronic structure of the active site, promoting the catalytic reaction. However, they typically require high noble metal loadings, whereas reducing the noble metal loading of the catalyst is essential to reduce costs. Efforts towards developing cost-effective, highly active, and selective catalysts for CO2RR are critical for the widespread adoption of this promising technology.
Recent studies by Choi et al. reported that electrochemical atomic layer deposition can be used to epitaxially grow an atomically thin Pt film on graphene ( ):11 this process not only provides bulk-like stability but also creates a unique electronic structure, where the
):11 this process not only provides bulk-like stability but also creates a unique electronic structure, where the  orbitals of Pt atoms are significantly decreased near the Fermi level due to a combination of interplanar Pt-carbon covalent bonding and inter/intraplanar metallic bonding. This newly discovered covalent bonding between the metal and graphene in 2D metal/graphene (
orbitals of Pt atoms are significantly decreased near the Fermi level due to a combination of interplanar Pt-carbon covalent bonding and inter/intraplanar metallic bonding. This newly discovered covalent bonding between the metal and graphene in 2D metal/graphene (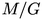 ) systems not only stabilizes the metal but also benefits from the intrinsic properties of graphene such as near-metallic conductivity and mechanical strength. Additionally, the properties can be transferred between the graphene and metal layers, as demonstrated by the “chemical transparency” of graphene-capped Pt catalysts that adopt catalytic properties at the graphene side of the underlying Pt.12 Furthermore, we can use the concept of “remote epitaxy”, introduced by Kim et al.,13 where the structure of films grown on one side of graphene is directed by the structure of the support on the reverse side. This concept is particularly relevant for catalytic applications, where epitaxy between metal catalysts and
) systems not only stabilizes the metal but also benefits from the intrinsic properties of graphene such as near-metallic conductivity and mechanical strength. Additionally, the properties can be transferred between the graphene and metal layers, as demonstrated by the “chemical transparency” of graphene-capped Pt catalysts that adopt catalytic properties at the graphene side of the underlying Pt.12 Furthermore, we can use the concept of “remote epitaxy”, introduced by Kim et al.,13 where the structure of films grown on one side of graphene is directed by the structure of the support on the reverse side. This concept is particularly relevant for catalytic applications, where epitaxy between metal catalysts and  carbon strains the catalyst and affects catalytic activity, as demonstrated for the oxygen reduction reaction (ORR).11b These findings provide insight into the potential for using
carbon strains the catalyst and affects catalytic activity, as demonstrated for the oxygen reduction reaction (ORR).11b These findings provide insight into the potential for using 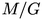 systems for catalytic applications and highlight the importance of understanding the interactions between metal catalysts and graphene supports.
systems for catalytic applications and highlight the importance of understanding the interactions between metal catalysts and graphene supports.
Building upon the paradigm set by the 2D  structure in electrocatalysis, we have expanded our investigation to include other late transition
structure in electrocatalysis, we have expanded our investigation to include other late transition  ,
,  , and
, and  metal/graphene structures. We have analyzed the formation of covalent bonding by examining the specific electronic structure of the metal surface. Our research reveals that the average energy position of interplanar
metal/graphene structures. We have analyzed the formation of covalent bonding by examining the specific electronic structure of the metal surface. Our research reveals that the average energy position of interplanar  orbitals (those with z components) near the Fermi level is of utmost importance. Moreover, we have discovered new structures where additional metal layers can be deposited on the graphene side of an
orbitals (those with z components) near the Fermi level is of utmost importance. Moreover, we have discovered new structures where additional metal layers can be deposited on the graphene side of an 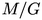 structure, creating graphene-sandwiched
structure, creating graphene-sandwiched  structures. We note here that chemical bond formation is enabled on the hybridized graphene side. For example, although copper does not form a chemical bond with pristine graphene, it does when deposited on the graphene side of
structures. We note here that chemical bond formation is enabled on the hybridized graphene side. For example, although copper does not form a chemical bond with pristine graphene, it does when deposited on the graphene side of  . Using these structures, we performed onset potential calculations of CO2RR to HCOOH as well as HER to evaluate true productivity and selectivity. Multiple experimental results later confirmed our computational findings. Our results indicate that the rational design of
. Using these structures, we performed onset potential calculations of CO2RR to HCOOH as well as HER to evaluate true productivity and selectivity. Multiple experimental results later confirmed our computational findings. Our results indicate that the rational design of  and
and  catalysts could lead to further improvements in the selective production of HCOOH.
catalysts could lead to further improvements in the selective production of HCOOH.
2 Results and Discussion
2.1 Structural Analysis of Metal /Graphene Structure
To fully understand the reaction kinetics on metal monolayer/graphene (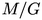 ) surfaces and to effectively tune CO2RR electrocatalytic activity and stability, it is crucial to have a clear understanding of the structure and composition of the
) surfaces and to effectively tune CO2RR electrocatalytic activity and stability, it is crucial to have a clear understanding of the structure and composition of the 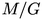 structure. In particular, the covalent bonding between the metal and graphene is a critical factor that can significantly impact the surface reactivity of 2D hybrid electrocatalysts. The electronic mixing between the metal and graphene can modify the electronic structure and activity of the surface, making it essential to scrutinize the electronic structure of the surface.
structure. In particular, the covalent bonding between the metal and graphene is a critical factor that can significantly impact the surface reactivity of 2D hybrid electrocatalysts. The electronic mixing between the metal and graphene can modify the electronic structure and activity of the surface, making it essential to scrutinize the electronic structure of the surface.
Figure 1a reveals that the bridge site yields the strongest covalent bonding between carbon and  and
and  transition metal atoms such as Ru, Rh, Pd, Ir, and Pt. In contrast, the ferromagnetic elements such as Fe, Co, and Ni prefer to adsorb in the on-top position of carbon as shown in Figure 1b. Group 11 elements such as Cu, Ag, and Au show strong van der Waals (vdW) interactions, with distances to graphene of 3.84 Å, 3.18 Å, and 3.59 Å, respectively (Figure S-4). To understand the various adsorption characteristics of these metals, we analyzed the local density of states (LDOS) to scrutinize the electronic structure of valence
transition metal atoms such as Ru, Rh, Pd, Ir, and Pt. In contrast, the ferromagnetic elements such as Fe, Co, and Ni prefer to adsorb in the on-top position of carbon as shown in Figure 1b. Group 11 elements such as Cu, Ag, and Au show strong van der Waals (vdW) interactions, with distances to graphene of 3.84 Å, 3.18 Å, and 3.59 Å, respectively (Figure S-4). To understand the various adsorption characteristics of these metals, we analyzed the local density of states (LDOS) to scrutinize the electronic structure of valence  orbitals of metal monolayer (denoted as
orbitals of metal monolayer (denoted as . As illustrated in Figure S-5, there is a distinct difference in the density of
. As illustrated in Figure S-5, there is a distinct difference in the density of  orbitals below the Fermi level, especially in the energy range,
orbitals below the Fermi level, especially in the energy range, 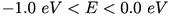 , among the physisorbed metals and the chemisorbed metals at the bridge-site and on-top position of graphene. For the bridge site-binding metals such as
, among the physisorbed metals and the chemisorbed metals at the bridge-site and on-top position of graphene. For the bridge site-binding metals such as 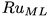 ,
,  ,
,  ,
,  , and
, and 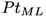 , both spin-up and spin-down
, both spin-up and spin-down  electron states exhibit a high density at
electron states exhibit a high density at 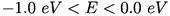 . In contrast, for the top site-adsorbed metals such as
. In contrast, for the top site-adsorbed metals such as  ,
,  , and
, and 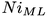 , the spin-down state is dominant in comparison to the spin-up state due to the spin asymmetry originating from magnetic properties. Physisorbed metals, such as
, the spin-down state is dominant in comparison to the spin-up state due to the spin asymmetry originating from magnetic properties. Physisorbed metals, such as 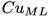 ,
, 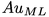 , and
, and  , exhibit distinct electronic characteristics. Please note that the energy levels near the Fermi level are practically unoccupied for both spin-up and spin-down states, indicating that the high and low density of states near the Fermi level play an essential role in the formation of strong and weak covalent bonds between metal and graphene, respectively.
, exhibit distinct electronic characteristics. Please note that the energy levels near the Fermi level are practically unoccupied for both spin-up and spin-down states, indicating that the high and low density of states near the Fermi level play an essential role in the formation of strong and weak covalent bonds between metal and graphene, respectively.

Schematic illustration of the geometry optimized structures: A) and B) Metal/Graphene (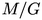 ), and C) Metal/Graphene/Metal (
), and C) Metal/Graphene/Metal ( ) structures used in this study. Metal adsorbs at A) bridge site or; B) on-top site of graphene to form
) structures used in this study. Metal adsorbs at A) bridge site or; B) on-top site of graphene to form 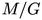 structures.
structures.
Although the metal LDOS provides general insight into strong/weak bond formation, it remains challenging to define a more descriptive bond formation mechanism with LDOS because the five  orbitals do not contribute equally to interatomic orbital hybridization. To unravel the role of specific
orbitals do not contribute equally to interatomic orbital hybridization. To unravel the role of specific  orbitals in interplanar metal–carbon
orbitals in interplanar metal–carbon  interactions, we further analyzed the orbital resolved density of states (ORDOS).
interactions, we further analyzed the orbital resolved density of states (ORDOS).
First, we attribute the distribution of several small peaks in 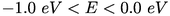 of
of  ,
, 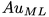 , and
, and  to the intraplanar orbitals
to the intraplanar orbitals  and
and  , which are mostly associated with metallic bonding. Considering the ligand positions in the geometry where
, which are mostly associated with metallic bonding. Considering the ligand positions in the geometry where  and
and  orbitals lie in the x–y plane (as shown in Figure S-6),
orbitals lie in the x–y plane (as shown in Figure S-6),  ,
,  , and
, and  orbitals are utilized for the interplanar bonding. However,
orbitals are utilized for the interplanar bonding. However,  ,
,  , and
, and  orbitals are completely unoccupied in the same energy range, making difficult to identify
orbitals are completely unoccupied in the same energy range, making difficult to identify  covalency in
covalency in  ,
, 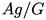 , and
, and  systems.
systems.
Second, for  and
and  metals such as
metals such as 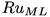 ,
, 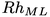 ,
,  ,
,  , and
, and 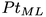 that bind at the bridge site of graphene, both spin up and down states in
that bind at the bridge site of graphene, both spin up and down states in  ,
,  , and
, and  orbitals exhibit higher electronic density than
orbitals exhibit higher electronic density than  and
and  , in general, indicating these interplanar orbitals are more responsible than the intraplanar orbitals for the
, in general, indicating these interplanar orbitals are more responsible than the intraplanar orbitals for the  covalent bonding (as shown in Figures S-7 and S-8).
covalent bonding (as shown in Figures S-7 and S-8).
Third, for ferromagnetic  metals such as
metals such as  ,
,  , and
, and 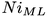 , spin-down states are solely present in
, spin-down states are solely present in 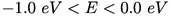 for
for  ,
,  , and
, and  orbitals while the spin-up states in the same energy range are found instead in the intraplanar orbitals
orbitals while the spin-up states in the same energy range are found instead in the intraplanar orbitals  and
and  (as shown in Figure S-9). Here, we discovered that the
(as shown in Figure S-9). Here, we discovered that the  orbital has a much higher peak density than
orbital has a much higher peak density than  and
and  orbitals, indicating that the contribution of the spin-down state in
orbitals, indicating that the contribution of the spin-down state in  orbital is the most influential to the formation of
orbital is the most influential to the formation of  bond at the top site of graphene.
bond at the top site of graphene.
The specific  orbitals involved in
orbitals involved in  covalent bonding formation are summarized in Figure 2. Typically, a single state at the
covalent bonding formation are summarized in Figure 2. Typically, a single state at the  -band center,
-band center,  , is used to approximate the band of d-states involved in the interaction and its correlation with the adsorption energy of the metal on graphene. In the case of metals (
, is used to approximate the band of d-states involved in the interaction and its correlation with the adsorption energy of the metal on graphene. In the case of metals (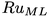 ,
,  ,
,  ,
,  , and
, and  ) that adsorb at the bridge site of graphene, the upshift of
) that adsorb at the bridge site of graphene, the upshift of  of
of  ,
,  , and
, and  orbitals in
orbitals in 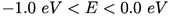 toward the Fermi level results in a stronger
toward the Fermi level results in a stronger  covalent bond. The strongest covalent bond is observed for
covalent bond. The strongest covalent bond is observed for 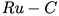 , followed by
, followed by 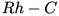 ,
,  ,
,  , and
, and 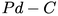 . For example,
. For example,  of
of 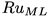 is the closest to the Fermi level, which lies at
is the closest to the Fermi level, which lies at  , followed by
, followed by  (
(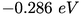 ),
),  (
(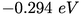 ),
), 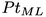 (
(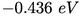 ), and
), and  (
( ), and the binding energy on graphene is the strongest for
), and the binding energy on graphene is the strongest for 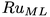 (
(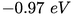 ), followed by
), followed by 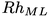 (
( ),
),  (
(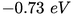 ),
), 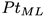 (
(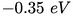 ), and
), and  (
(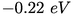 ). However, the ferromagnetic elements
). However, the ferromagnetic elements  ,
,  , and
, and 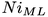 are weakly adsorbed on graphene compared to the
are weakly adsorbed on graphene compared to the  and
and  metals due to the downshift of
metals due to the downshift of  of
of  orbitals in
orbitals in 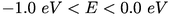 away from the Fermi level, with
away from the Fermi level, with  values of
values of  ,
,  , and
, and  , respectively. To summarize, not all d orbitals equally participate in the
, respectively. To summarize, not all d orbitals equally participate in the  bond formation, but rather the orbitals in the Z-directions within
bond formation, but rather the orbitals in the Z-directions within 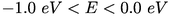 play a decisive role in determining the strength of the
play a decisive role in determining the strength of the  covalency.
covalency.

The effect of the average position of various  orbitals (
orbitals ( ) on the binding energy of
) on the binding energy of  on the graphene. Black circles, green squares, and red hexagons represent the
on the graphene. Black circles, green squares, and red hexagons represent the  orbital in the entire energy range,
orbital in the entire energy range,  ,
,  , and
, and  combined in
combined in 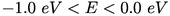 , and
, and  in
in 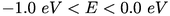 , respectively.
, respectively.
2.2 Change in Electronic Structure of Metal/Graphene Structure
The establishment of strong  covalency via
covalency via 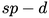 orbital hybridization results in a significant modification of the electronic structure of the metal surface. Specifically, we observe a remarkable reduction of the
orbital hybridization results in a significant modification of the electronic structure of the metal surface. Specifically, we observe a remarkable reduction of the  orbital LDOS near the Fermi level, particularly in
orbital LDOS near the Fermi level, particularly in 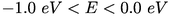 for the strongly chemisorbed metals such as
for the strongly chemisorbed metals such as  ,
,  ,
,  ,
,  , and
, and 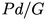 , compared to pure
, compared to pure  (Figure S-10). This reduction in electron density is driven by charge transfer from metal to graphene, as indicated by the Bader charge analysis in Table S-2. In contrast, due to weaker
(Figure S-10). This reduction in electron density is driven by charge transfer from metal to graphene, as indicated by the Bader charge analysis in Table S-2. In contrast, due to weaker  covalency, the interatomic electronic mixing between ferromagnetic metals and graphene leads to relatively minor modification in the
covalency, the interatomic electronic mixing between ferromagnetic metals and graphene leads to relatively minor modification in the  orbital LDOS. For instance, in
orbital LDOS. For instance, in 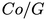 , there is no significant downshift or upshift of
, there is no significant downshift or upshift of  orbitals for spin-up and -down states, and only a slight decrease of LDOS within
orbitals for spin-up and -down states, and only a slight decrease of LDOS within 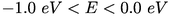 . Similarly, for
. Similarly, for 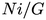 the reduction of the spin-down state is greater in the same range, which may be attributed to the greater amount of charge transferred (
the reduction of the spin-down state is greater in the same range, which may be attributed to the greater amount of charge transferred (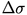 ) to graphene for
) to graphene for 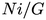 (
( ) than for
) than for 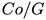 (
( ). These changes in the
). These changes in the  orbitals will be further discussed in relation to the binding energy of HCOO in Section 3.4.
orbitals will be further discussed in relation to the binding energy of HCOO in Section 3.4.
2.3 Structural Analysis of Metal/Graphene/Metal Structure
The electronic structure of metal surfaces in 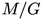 can be further modified by depositing an additional metal film on the graphene side, creating
can be further modified by depositing an additional metal film on the graphene side, creating  structures, as shown in Figure 1C. In a previous study, Choi et al. discovered that electron density accumulation at the
structures, as shown in Figure 1C. In a previous study, Choi et al. discovered that electron density accumulation at the  bond could facilitate the formation of additional
bond could facilitate the formation of additional  covalent bonding.11
covalent bonding.11
In this study, we deposited 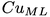 ,
,  ,
,  , and
, and 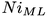 , identified as either physisorbed or weakly adsorbed to graphene, on the graphene side of
, identified as either physisorbed or weakly adsorbed to graphene, on the graphene side of  ,
,  ,
,  ,
,  ,
, 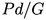 ,
, 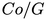 ,
, 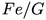 , and
, and 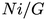 structures. Surprisingly,
structures. Surprisingly, 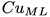 , once physisorbed on pristine graphene, was found to be chemisorbed on hybridized graphene in
, once physisorbed on pristine graphene, was found to be chemisorbed on hybridized graphene in  ,
, 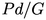 ,
,  , and
, and  structures. The binding energy of
structures. The binding energy of 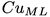 became as strong as
became as strong as  per
per  atom on
atom on  , followed by
, followed by 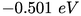 per
per  atom on
atom on 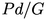 ,
, 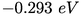 per
per  atom on
atom on  , and
, and 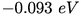 on
on  structures. Similarly,
structures. Similarly,  ,
,  , and
, and 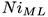 formed stronger chemical bonding on the hybridized graphene in
formed stronger chemical bonding on the hybridized graphene in 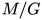 structures than on the pristine graphene.
structures than on the pristine graphene.
To validate our computational models, we attempted electrochemical deposition of  under the same conditions directly onto graphene and onto graphene supported by
under the same conditions directly onto graphene and onto graphene supported by  . As shown in Figure 3, we found impossible to synthesize
. As shown in Figure 3, we found impossible to synthesize 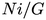 , but we could synthesize
, but we could synthesize 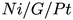 with the help of
with the help of  on the reverse side of graphene.
on the reverse side of graphene.

Experimental validation 
 XPS using cyclic voltammetry that
XPS using cyclic voltammetry that  will not directly deposit on graphene (upper left), but will do so (upper right), if
will not directly deposit on graphene (upper left), but will do so (upper right), if  is present on the other side of graphene.This validates our predictions.
is present on the other side of graphene.This validates our predictions.
To gain a deeper understanding of the atomic-level formation of  structures, we analyze the electronic structure of graphene in
structures, we analyze the electronic structure of graphene in 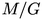 structures, where the graphene serves as an active site in this context. Due to
structures, where the graphene serves as an active site in this context. Due to 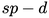 orbital hybridization, the
orbital hybridization, the  orbitals of graphene in
orbitals of graphene in 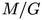 structures undergo significant changes in comparison to pristine graphene, as depicted in Figure 4. The most notable difference is the appearance of a sharp electron density peak near the Fermi level, especially between
structures undergo significant changes in comparison to pristine graphene, as depicted in Figure 4. The most notable difference is the appearance of a sharp electron density peak near the Fermi level, especially between 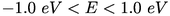 , which results from charge transfer from metal to graphene. We have confirmed that the newly generated peaks are exclusively attributed to this additional charge in the graphene, rather than strain imposed by the metal. The electronic structure of the hybridized graphene is modified not only by the mismatch of lattice parameters between metal and graphene (known as the strain effect), but also by the charge polarization induced by the heteronuclear interactions between metal and carbon atoms (known as the ligand effect). To isolate the contributions of the strain and ligand effects from the change in electronic structure, we obtained the strain contribution by manipulating the lattice parameter of pure graphene corresponding to
, which results from charge transfer from metal to graphene. We have confirmed that the newly generated peaks are exclusively attributed to this additional charge in the graphene, rather than strain imposed by the metal. The electronic structure of the hybridized graphene is modified not only by the mismatch of lattice parameters between metal and graphene (known as the strain effect), but also by the charge polarization induced by the heteronuclear interactions between metal and carbon atoms (known as the ligand effect). To isolate the contributions of the strain and ligand effects from the change in electronic structure, we obtained the strain contribution by manipulating the lattice parameter of pure graphene corresponding to 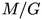 structures and calculating their electronic structures, as shown in Figure S-11. Regardless of the level of strain, the shape of the
structures and calculating their electronic structures, as shown in Figure S-11. Regardless of the level of strain, the shape of the  band for strained graphene remains very similar to that of pristine graphene, with only an upshift of the average position of the
band for strained graphene remains very similar to that of pristine graphene, with only an upshift of the average position of the  orbital (denoted as
orbital (denoted as  ) towards the Fermi level.
) towards the Fermi level.

The projected density of states on the  (red) and
(red) and  (black) band of graphene in
(black) band of graphene in 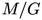 structures (denoted as
structures (denoted as  ) and pristine graphene shown in solid and dotted line, respectively. The dotted vertical line at 0 eV corresponds to the Fermi level.
) and pristine graphene shown in solid and dotted line, respectively. The dotted vertical line at 0 eV corresponds to the Fermi level.
Since not all three  orbitals of the graphene in
orbitals of the graphene in 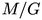 equally contribute to the formation of
equally contribute to the formation of  structures, we performed ORDOS analysis to decompose the
structures, we performed ORDOS analysis to decompose the  orbitals of graphene in the
orbitals of graphene in the 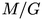 structures, as shown in Figure S-12. This analysis revealed that the electron density peaks generated near the Fermi level primarily arise from the intraplanar
structures, as shown in Figure S-12. This analysis revealed that the electron density peaks generated near the Fermi level primarily arise from the intraplanar 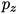 orbital, while no significant electron density was found in the
orbital, while no significant electron density was found in the  and
and  orbitals in this energy range. Therefore, we determined that the intraplanar
orbitals in this energy range. Therefore, we determined that the intraplanar 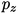 orbital in the valence region at
orbital in the valence region at 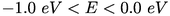 plays a crucial role in the adsorption of additional metal monolayers on graphene side of
plays a crucial role in the adsorption of additional metal monolayers on graphene side of 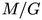 structures.
structures.
We then quantified the area under the spin up and down ( ) of the
) of the 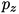 band at
band at 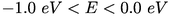 and correlated it with the binding energy of
and correlated it with the binding energy of  ,
,  ,
,  , and
, and 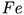 as shown in Figure 5. Our results showed that as
as shown in Figure 5. Our results showed that as  increases, the binding energy of additional metal on graphene tends to increase as well. Specifically,
increases, the binding energy of additional metal on graphene tends to increase as well. Specifically,  had the highest
had the highest  value of
value of  , exhibiting the strongest binding energies, followed by
, exhibiting the strongest binding energies, followed by  (
( ),
), 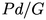 (
( ),
),  (
( ),
), 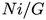 (
( ), and
), and 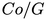 (
( ). Although
). Although  for
for 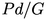 is slightly higher than
is slightly higher than  , the reason
, the reason  shows the stronger binding energy is that the average position of the
shows the stronger binding energy is that the average position of the 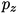 band is farther from the Fermi level for
band is farther from the Fermi level for  (
( ) than for
) than for 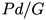 (
( ), which is analogous to the
), which is analogous to the  -band center theory.
-band center theory.

Binding energy of  ,
,  ,
,  , and
, and 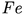 on the hybridized graphene of various
on the hybridized graphene of various 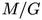 structures with respect to the area density of
structures with respect to the area density of  orbital in the energy range of −1 to 0 eV.
orbital in the energy range of −1 to 0 eV.
Therefore, we can conclude that the role of  of the
of the 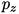 band at
band at  is critical in the formation of
is critical in the formation of  structures. Additionally, the average position of the
structures. Additionally, the average position of the  orbital in the same energy range should also be considered if
orbital in the same energy range should also be considered if  values are similar.
values are similar.
2.4 Electrochemical CO2RR to HCOOH on 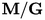 and
and  structures
structures
 (1)
(1) (2)
(2) (3)
(3)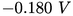 vs reversible hydrogen electrode (RHE).14 In aqueous electrolytes, the HER inevitably takes place via the following sequential steps15 which competes with the CO2RR:
vs reversible hydrogen electrode (RHE).14 In aqueous electrolytes, the HER inevitably takes place via the following sequential steps15 which competes with the CO2RR:
 (4)
(4) (5)
(5) (6)
(6)To promote selectivity toward HCOOH, the unwanted side reactions should be suppressed. Because HER is a very fast reaction with essentially no overpotential, it is quite challenging to find a catalyst that performs CO2RR preferentially at a reasonable overvoltage.
To evaluate the global activity and selectivity of CO2RR to HCOOH on all studied 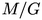 and
and  catalysts, we calculated the free energy changes in the two-step CO2RR, and the limiting potential at which all the elementary reaction steps become exergonic was obtained with respect to the free energy of HCOO binding energy (
catalysts, we calculated the free energy changes in the two-step CO2RR, and the limiting potential at which all the elementary reaction steps become exergonic was obtained with respect to the free energy of HCOO binding energy ( ), since HCOO is the only intermediate specie in CO2RR to HCOOH, formed as a result of CO2 protonation. Since HCOO* governs both Equations (1) and (2), it is reasonable to set
), since HCOO is the only intermediate specie in CO2RR to HCOOH, formed as a result of CO2 protonation. Since HCOO* governs both Equations (1) and (2), it is reasonable to set  as a descriptor.16 Properly tailoring the electronic structure of the active site to acquire moderate HCOO binding energy is necessary, as too strong or too weak binding energy of HCOO results in high overpotential.
as a descriptor.16 Properly tailoring the electronic structure of the active site to acquire moderate HCOO binding energy is necessary, as too strong or too weak binding energy of HCOO results in high overpotential.
In general, the deposition of extra metal on the hybridized graphene in  structures induces weaker interactions with the HCOO species in comparison to
structures induces weaker interactions with the HCOO species in comparison to 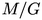 structures. Unlike the traditional
structures. Unlike the traditional  -band center theory, where the upward shift of the
-band center theory, where the upward shift of the  -band center relative to the Fermi level presents more binding affinity to adsorbate, the
-band center relative to the Fermi level presents more binding affinity to adsorbate, the  -band center of
-band center of  structures exhibits the opposite trend (as shown in Figure S-13). The
structures exhibits the opposite trend (as shown in Figure S-13). The  -band center theory is limited to the
-band center theory is limited to the  orbital hybridization, and the simple
orbital hybridization, and the simple  -band center correlation does not hold for the adsorption of HCOO on the
-band center correlation does not hold for the adsorption of HCOO on the 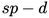 hybridized metal surface.17 Instead, it is the specific
hybridized metal surface.17 Instead, it is the specific  orbital in the specific energy range that determines the binding affinity to HCOO. In a previous study, it was revealed that
orbital in the specific energy range that determines the binding affinity to HCOO. In a previous study, it was revealed that  and
and  orbitals govern the chemisorption to bidentate HCOO specie, and as the density of these orbitals near the Fermi level decreases, the binding energy of HCOO tends to show an ascending trend.18 Similarly, the density of
orbitals govern the chemisorption to bidentate HCOO specie, and as the density of these orbitals near the Fermi level decreases, the binding energy of HCOO tends to show an ascending trend.18 Similarly, the density of  and
and  combined of
combined of 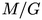 and
and  structures between
structures between  1.0
1.0 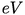 and 0.0
and 0.0 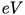 is correlated with the binding strength to HCOO.
is correlated with the binding strength to HCOO.
For example, Figure 6 shows the scaling relation between the adsorption energy of HCOO* and the density of  and
and  on various
on various 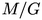 and
and  structures. Although there is a general consensus that the higher density of
structures. Although there is a general consensus that the higher density of  and
and  leads to a stronger interaction with HCOO*,
leads to a stronger interaction with HCOO*,  cases tend to deviate from the scaling relation due to the coupling interaction between the metal
cases tend to deviate from the scaling relation due to the coupling interaction between the metal  orbital and carbon
orbital and carbon  orbital, broadening of
orbital, broadening of  -bandwidth of
-bandwidth of  , and an upsurge in
, and an upsurge in  density. Thus, the increased
density. Thus, the increased  and
and  density in
density in 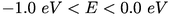 , which are contradictory to the studied structures. Consequently, the density of
, which are contradictory to the studied structures. Consequently, the density of  and
and  may not be the only indicator to the binding energies of HCOO*, and a more precise approach is perhaps required for future studies.
may not be the only indicator to the binding energies of HCOO*, and a more precise approach is perhaps required for future studies.

Binding energy of HCOO on 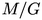 and
and 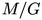 /M structures with respect to the area density of
/M structures with respect to the area density of  orbitals combined in
orbitals combined in 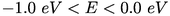 .
.
Figure 7 shows that  has the highest limiting potential of
has the highest limiting potential of 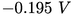 vs. RHE, which is closest to the calculated equilibrium potential at
vs. RHE, which is closest to the calculated equilibrium potential at 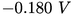 vs. RHE. This indicates that the overpotential is the smallest for this catalyst, followed by
vs. RHE. This indicates that the overpotential is the smallest for this catalyst, followed by  ,
,  ,
, 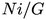 ,
,  , and
, and 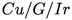 . Among
. Among 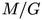 structures,
structures,  and
and  are active toward HCOOH production. However,
are active toward HCOOH production. However, 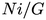 exhibits high HCOOH productivity, but the adsorption of HCOO destabilizes the
exhibits high HCOOH productivity, but the adsorption of HCOO destabilizes the  surface due to poor covalency between
surface due to poor covalency between  and graphene. Similar structural deformation is observed for
and graphene. Similar structural deformation is observed for 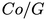 and
and 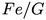 . Such deformation is not found in
. Such deformation is not found in  ,
,  , and
, and  structures, where the binding energy of HCOO is generally stronger than in their respective
structures, where the binding energy of HCOO is generally stronger than in their respective 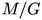 counterparts. Consequently, the limiting potentials for
counterparts. Consequently, the limiting potentials for  shift up, while for
shift up, while for  , and
, and  they shift down.
they shift down.

Calculated limiting potential (UL) for CO2RR to HCOOH as a function of the free energy of HCOO adsorption for the studied catalysts. Red triangle symbol denotes unstable structures in which metals are detached from the graphene under the presence of HCOO on the surface.
It is crucial to evaluate the competing HER for selectivity since the equilibrium potential for HER is more positive than that for CO2RR to HCOOH. As a result,  ,
,  , and
, and 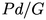 electrocatalysts have limiting potentials (UL) that are more positive than that of HER, as shown in Figure S-14. This is surprising, as pure monometallic
electrocatalysts have limiting potentials (UL) that are more positive than that of HER, as shown in Figure S-14. This is surprising, as pure monometallic  nanoparticles are known to be excellent catalysts for HER. In addition, we considered CO2RR to CO formation via COOH*. According to the free energy profile in Figure S-15, for
nanoparticles are known to be excellent catalysts for HER. In addition, we considered CO2RR to CO formation via COOH*. According to the free energy profile in Figure S-15, for  at −0.29 V, the UL for CO formation, CO2RR to HCOOH pathway remains endergonic while the CO pathway becomes exergonic, indicating a preference for CO production. On the other hand, we found that
at −0.29 V, the UL for CO formation, CO2RR to HCOOH pathway remains endergonic while the CO pathway becomes exergonic, indicating a preference for CO production. On the other hand, we found that 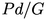 and
and  are more selective toward HCOOH formation over CO.
are more selective toward HCOOH formation over CO.
As a validation of the theoretical predictions, we experimentally assessed the electrochemical onset potential for HCOOH formation, as illustrated in Figure S-16. Although the onset potential for  could not be obtained, presumably due to a more positive UL for CO than HCOOH,
could not be obtained, presumably due to a more positive UL for CO than HCOOH, 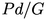 and
and  exhibit the highest onset potentials followed by
exhibit the highest onset potentials followed by  ,
,  , and
, and  . Despite a marginal computational prediction favoring HER over CO2RR to HCOOH in
. Despite a marginal computational prediction favoring HER over CO2RR to HCOOH in  by 0.021 V, the experimental data showed a higher selectivity toward HCOOH, possibly due to the intrinsic error in DFT calculations. The
by 0.021 V, the experimental data showed a higher selectivity toward HCOOH, possibly due to the intrinsic error in DFT calculations. The  and
and  cases were chosen to verify the computational prediction that these structural inverses should have similar reaction potential, while
cases were chosen to verify the computational prediction that these structural inverses should have similar reaction potential, while  was chosen because it is predicted to have a significantly lower potential than the first two. The computational model predictions align well with the experimental data, confirming the order and similarity in onset potentials for
was chosen because it is predicted to have a significantly lower potential than the first two. The computational model predictions align well with the experimental data, confirming the order and similarity in onset potentials for  and
and  with
with 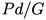 and
and  exhibiting a higher and lower potential, respectively.
exhibiting a higher and lower potential, respectively.
This 2D hybrid catalyst design methodology is not limited to CO2RR but can also be applied to other electrochemical reactions, such as nitrogen reduction reaction, which is still challenging with conventional metallic electrocatalysts.
3 Conclusion
With increasing environmental and energy concerns, there is a pressing need for the development of effective and affordable methods to convert atmospheric CO2 into valuable fuels. This study employed theoretical methods, with experimental validation, to investigate the electrochemical reduction of CO2 to HCOOH on 2D hybrid metal/graphene and metal/graphene/metal structures. The charge transfer from metal to graphene in the metal/graphene structures allows for electrodeposition of additional metal thin films, resulting in enhanced catalytic activity. We have identified the necessary requirements for catalyst materials to be suitable for highly active and selective CO2RR to HCOOH over CO formation and HER. Our study reveals that a high (low) density of  and
and  orbitals near the Fermi level leads to a strong (weak) interaction with HCOO, providing a key factor in designing highly active CO2RR electrocatalysts. Our results show that
orbitals near the Fermi level leads to a strong (weak) interaction with HCOO, providing a key factor in designing highly active CO2RR electrocatalysts. Our results show that 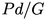 ,
,  , and
, and  are highly selective toward HCOOH production among various metal/graphene and metal/graphene/metal structures. Our
are highly selective toward HCOOH production among various metal/graphene and metal/graphene/metal structures. Our  orbital driven 2D hybrid catalyst design methodology is widely applicable not only to CO2RR but also to other thermochemical and electrochemical reactions, providing opportunities for the establishment of novel catalysts and optimization of catalytic performance through electronic mixing of metal and other
orbital driven 2D hybrid catalyst design methodology is widely applicable not only to CO2RR but also to other thermochemical and electrochemical reactions, providing opportunities for the establishment of novel catalysts and optimization of catalytic performance through electronic mixing of metal and other  -block elements.
-block elements.
Supporting Information
The authors have cited additional references within the Supporting Information.19, 20, 21-24, 25
Acknowledgments
The work performed by WAG was supported by the Liquid Sun-light Alliance, which is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, Fuels from Sunlight Hub under Award DE-SC0021266.
Conflict of interests
The authors declare no conflict of interest.
Open Research
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.

