

CycloSiFA: The Next Generation of Silicon-Based Fluoride Acceptors for Positron Emission Tomography (PET)
Graphical Abstract
Cyclic azasilole derivatives undergo ring-opening with 18F– anions under mild conditions and with high radiochemical yield. They represent a novel class of Silicon Fluoride Acceptor systems (CycloSiFA) and hold great potential for convenient kit-like labelling procedures in different medicinal applications.

Abstract
The ring-opening Si-fluorination of a variety of azasilole derivatives cyclo-1-(iPr2Si)−4-X−C6H3−2-CH2NR (4: R=2,6-iPr2C6H3, X=H; 4 a: R=2,4,6-Me3C6H2, X=H; 9: R=2,6-iPr2C6H3, X=tBuMe2SiO; 10: R=2,6-iPr2C6H3, X=OH; 13: R=2,6-iPr2C6H3, X=HCCCH2O; 22: R=2,6-iPr2C6H3, X=tBuMe2SiCH2O) with different 19F-fluoride sources was studied, optimized and the experience gained was used in a translational approach to create a straightforward 18F-labelling protocol for the azasilole derivatives [18F]6 and [18F]14. The latter constitutes a potential clickable CycloSiFA prosthetic group which might be used in PET tracer development using Cu-catalysed triazole formation. Based on our findings, CycloSiFA has the potential to become a new entry into non-canonical labelling methodologies for radioactive PET tracer development.
Introduction
The advent of non-canonical radio-fluorine (fluorine-18 isotope, 18F, half-life=109.7 min) labelling chemistries based on silicon-, boron-, aluminium-18F formation and the most recently reported aryl fluorosulfate (SuFEx) click radiolabelling has introduced new synthetic tools to radiopharmaceutical chemistry. It facilitated the development of molecular imaging agents for Positron Emission Tomography (PET).1, 2, 3-6
PET is a diagnostic imaging modality based on the detection of gamma rays originating from positron electron annihilation.7, 8 The positron is the decay product of positron emitting radioisotopes (e.g. 18F). To obtain clinically useful radiotracers for in vivo imaging the 18F-isotope has to be stably bound to the imaging agent. Since radiochemists have to combat the radioactive decay, the introduction of 18F has to be swift and the workup procedures streamlined. 18F-PET tracer development has previously been limited mostly to carbon-18F bond chemistry,9, 10 characterized by the use of high temperatures, side product formation and sometimes long reaction times. Structurally complex molecules (peptides and proteins) could only be labelled by synthesizing small 18F-labelled prosthetic groups subsequently being attached to the complex molecules.11 Complex radiochemistry and laborious work up procedures hindered the dissemination of structurally intricate 18F-imaging agents.
Shifting away from the classical 18F-carbon bond radiochemistry, alternative elements such as boron,12 aluminium,13 silicon and most recently sulphur,14 all forming strong chemical bonds with fluorine, were employed to bind the 18F-isotope in one single step to complex molecules bearing a multitude of unprotected chemical groups. These one-step 18F-labelling protocols constitute a technical improvement over a multistep synthetic approach. The boron- and silicon 18F-chemistries additionally demonstrated that high molar activities Am (radioactivity/mol radiotracer) can be reached by isotopic exchange (IE) reactions where a non-radioactive fluorine (19F isotope), connected to a boron or silicon atom, is simply replaced by radioactive 18F. The most recently introduced SuFEx labelling approach confirmed these findings. The amount of precursor (fluoroborates, fluorosiliconates and fluorosulfates) can be reduced to nano-molar quantities, a prerequisite to achieve high Am formerly not possible in IE carbon-18F chemistry. Aluminium-18F chemistry capitalizes on a different labelling Scheme by preforming [Al−F] aqua complexes which can be further react with radiometal chelators.15
The Al-, B-, and Si-based 18F-radiochemistries have proven their clinical applicability and became already valuable additions to the portfolio of available radiotracers for PET imaging of neuroendocrine tumours,16-20 prostate specific membrane antigen (PSMA)21 and metastatic lung cancer.22a Previously disclosed silicon fluoride acceptors (SiFAs)2a can be considered being the first generation of Si-based 18F-radiopharmaceuticals (Scheme 1). [18F]SiTATE labelling has been clinically consolidated in more than 3000 cancer patients (several clinics in Germany) for the diagnosis of neuro-endocrine tumours and most recently for meningioma imaging.22b

18F-radiolabelling reactions using isotope exchange equilibrium with [19F]SiFAs or ring opening reaction of CycloSiFAs.
SiFAs are limited to IE reactions because any other group than F e.g. Cl, Br or I connected to the Si centre yields a chemically unstable precursor compound that cannot be further processed or derivatized for 18F-radiolabelling. Although the SiFA chemistry yields relatively high Am via IE, a direct labelling approach based on leaving group substitution might be advantageous by circumventing limitations imposed by the IE equilibrium to reach highest radiochemical conversions. To extend the field of Si-based fluorination chemistry and to devise an alternative leaving-group based 18F-labelling protocol, herein we introduce CycloSiFA, the first in kind cyclic labelling precursor amenable towards 18F-radiofluorination via Si−N ring opening reaction. CycloSiFA like the original SiFA can only be used for the 18F-labeling of large organic molecules such as peptides and proteins. The change of lipophilicity, by introducing SiFA, for small biomolecules would be too drastic and negatively impact the tracer's biodistribution. The calculated logP value of CycloSiFA of 7.3 stipulates the addition of hydrophilic auxiliaries (e.g. aspartic acids) to the biomolecule to be labelled to reduce the compound's overall lipophilicity, a crucial parameter for successful PET probe development. This strategy has been proven applicable to previously labelled SiFA compounds such a [18F]SiTATE, a compound for clinical somatostatin receptor imaging in cancer patients.19
Results and Discussion
The literature-known model compound 2-(2,6-diisopropylphenyl)-1,1-diphenyl-2,3-dihydro-1H-benzo[c][1,2]azasilole, 2, was synthesized according to Jambor et al. via halogen-metal exchange between o-bromobenzyldiisopropylphenyl imine, o-BrC6H4C(H)NC6H3-[CH(CH3)2]2–2,5, 1, and n-butyl lithium, nBuLi, followed by substitution with diphenylsilane, Ph2SiH2 (Scheme 2). The formation of the Si−N-bond is the result of spontaneous hydrosilylation of the CH=N imine moiety.23

Synthesis of 2-(2,6-diisopropylphenyl)-1,1-diphenyl-2,3-dihydro-1H-benzo[c][1,2]azasilole 2 and its reaction with hydrogen fluoride, HF. Commercial aqueous HF with concentration of 48–51 % was used.
Attempts to obtain the desired SiFA compound by treatment of 2 with potassium fluoride gave no conversion, whereas reaction with tetrabutylammonium fluoride, nBu4NF, hereafter referred to as TBAF, gave a complex reaction mixture which was not further analysed. Likewise, ring opening of the cyclic compound using commercial conc. hydrofluoric acid (48–51 %) afforded no fluorinated product. Rather, the silanol 3 was isolated, indicating a low hydrolytic stability of the Si−F-bond as previously reported by Ametamey et al. for similar compounds.24 In order to improve the hydrolytic stability, sterically more demanding groups needed to be attached to the silicon atom.
Classical SiFA compounds use tert-butyl substituents to shield the Si−F-bonds from hydrolytic degradation. The use of di-tert-butylsilane in the reaction shown above gave no substitution product, as did the application of Ir-catalysts for C−H-bond-activation,25a the use of directed ortho-metalation or a synthesis pathway via protected amines.
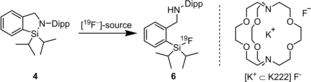
Since fluorodiisopropylphenylsilanes bearing a methyl group in ortho-position to the silicon substituent were reported to have good hydrolytic stability (half-life 302 h), diisopropylsilane was chosen as the reactant to afford the sterically more demanding CycloSiFA compound 4.24 The transformation of 1 with nBuLi and iPr2SiH2, however, gave 2-(2,6-diisopropylphenyl)-1,1-diisopropyl-2,3-dihydro-1H-benzo[c][1,2]azasilole, 4, with a poor yield of 8 % after 11 d reaction time (Scheme 3).

Optimized synthesis and molecular structure of 2-(2,6-diisopropylphenyl)-1,1-diisopropyl-2,3-dihydro-1H-benzo[c][1,2]azasilole 4 (CycloSiFA).
The use of THF instead of diethyl ether did not accelerate the reaction. To optimize the yield, the reaction was divided into two separate steps. First, the lithiation was conducted using nBuLi and diisopropylchlorosilane to afford the intermediate silane 5 in 91 % yield. Then, the use of the strong Lewis acid B(C6F5)3 enabled cyclization to CycloSiFA 4 in 86 % yield. Structure verification was accomplished by single crystal X-ray diffraction analysis25b (for details see the Supporting Information). CycloSiFA 4 shows excellent stability at room temperature, even in solution. However, due to its acid sensitivity, purification by column chromatography needed to be performed with neutral or basic aluminium oxide to prevent ring opening to the corresponding silanol. CycloSiFAs with different substituents at the nitrogen atom were prepared in lower yields by this route but they showed lower hydrolytic stability.26
The optimization for the 19F-fluorination of CycloSiFA 4 giving SiFA 6 was performed at room temperature in CH2Cl2. The use of 2.0 molar equivalents of conc. hydrofluoric acid or HF⋅py showed quantitative conversion to the desired product after just 10 min (Table 1, entries 1, 2).
|
|||||
Entry |
[19F−1]-Source |
Estimated concentration |
Additive |
t |
Yield [%][b] |
|---|---|---|---|---|---|
1 |
HF (aq., 2.0 equiv) |
c(Si)≈50 mM, c(F)≈100 mM |
|
10 min |
quantitative |
2 |
HF⋅py (2.0 equiv) |
c(Si)≈100 mM, c(F)≈200 mM |
|
10 min |
quantitative |
3 |
Et3N⋅3HF (3.0 equiv) |
c(Si)≈95 mM, c(F)≈286 mM |
|
2 h |
95 |
4[c] |
BF3⋅OEt2 (4.0 equiv) |
c(Si)≈95 mM, c(F)≈381 mM |
|
2 h |
quantitative |
5[d] |
TBAF[e] (1.0 equiv) |
c(Si)≈67 mM, c(F)≈67 mM |
Oxalic acid[f] |
90 min |
66 |
6[d] |
TBAF[e] (1.0 equiv) |
c(Si)≈77 mM, c(F)≈77 mM |
Acetic acid[f] |
3 h |
n. c. |
7[d] |
TBAF[e] (1.0 equiv) |
c(Si)≈91 mM, c(F)≈91 mM |
|
23 h |
n. c. |
8[d] |
[K+⊂ K222] F– (2.0 equiv) |
c(Si)≈63 mM, c(F)≈125 mM |
Oxalic acid[f] |
2 h |
n. c. |
9[d] |
[K+⊂ K222] F– (2.0 equiv) |
c(Si)≈63 mM, c(F)≈125 mM |
Acetic acid[f] |
22 h |
n. c. |
10[d] |
[K+⊂ K222] F– (2.0 equiv) |
c(Si)≈98 mM, c(F)≈196 mM |
Acetic acid[g] |
20 h |
n. c. |
11[d] |
[K+⊂ K222] F– (1.0 equiv) |
c(Si)≈100 mM, c(F)≈200 mM |
|
20 h |
n. c. |
- [a] Reactions at 22 °C in dry CH2Cl2. [b] Yield of isolated product. [c] At 30 °C. [d] Sonication for 5 min after mixing. [e] 1 m in THF. [f] 0.5 m in H2O. [g] Glacial acetic acid. n. c.=no conversion.
In the case of radiofluorination with 18F, this would imply a quantitative formation of the radiotracer, while classical SiFA compounds can only reach a theoretic maximum of 67 % due to the isotopic exchange equilibrium. Triethylamine trihydrofluoride (3.0 equiv) required a longer reaction time of 2 h to give a good yield of 95 % (entry 3). Boron trifluoride diethyl etherate (4.0 equiv) at 30 °C was also able to convert the substrate in quantitative yield after 2 h (entry 4).
Even after sonication and prolonged reaction times, the use of TBAF without an additive or with acetic acid did not yield any product (entries 6, 7). The addition of oxalic acid enabled product formation with 66 % yield after 90 min (entry 5). Such reaction conditions are known to increase the reactivity and are also applied in the radiofluorination with 18F.19, 20
During this optimization study, potassium fluoride and kryptofix 222 did not generate the fluorinated product 6. The addition of weak diluted acids or glacial acetic acid did not change the outcome (entries 8–11). The optimization showed that fluorination of CycloSiFA model compound 4 could be achieved not only using HF-containing reagents but also classical radiochemical labelling conditions. Moreover, unlike the diphenyl derivative 2, the stability of 4 against TBAF in the presence of water enables the synthesis of substituted CycloSiFAs for use in imaging applications.
The synthesis of the phenolic CycloSiFA derivative 10 was performed starting with the imine formation by condensation of the known aldehyde 727 and 2,6-diisopropylphenyl amine (Dipp-amine, Scheme 4).

Synthesis of the CycloSiFA 10 containing a hydroxy substituent at the aryl backbone and its deprotection/ring opening.
The subsequent halogen-metal exchange was performed using tBuLi instead of nBuLi since the reaction proceeded faster, and the progress of the reaction could be monitored visually because of a strong colour change to yellow and formation of a precipitate. Reaction of the lithiated imine with diisopropylchlorosilane gave the intermediate 8 in 75 % yield over two steps. Cyclization to the tert-butyldimethylsilyl-protected (hereafter referred to as TBS-protected) CycloSiFA 9 in the presence of B(C6F5)3 proceeded smoothly (94 % yield). This compound showed a unique reactivity towards different fluoride sources. The use of TBAF in THF enabled deprotection of the silyl ether to give the free phenol 10 after 40 min with 92 % yield. The Si−N-bond remained intact and the overall yield from 7 to 10 was 65 % over four steps. The phenol 10 could further be converted into the opened SiFA 11 by reaction with commercial hydrofluoric acid (48–51 %). This compound could also be obtained directly from 9 in quantitative yield by treatment with 6.0 equiv. of conc. hydrofluoric acid for 18 h at room temperature. However, if 9 was reacted with only 2.0 equiv. of conc. HF for 20 min, the Si−N-bond was cleaved but the silyl ether remained intact to afford 12 with quantitative yield.
The structure in the solid state (for details see the Supporting Information) of CycloSiFA 10 was confirmed by single crystal X-ray diffraction analysis.25b It shows the presence of the phenolic oxygen atom but in contrast to the structure of phenol28a-28d no intermolecular hydrogen bond. This was verified by an infrared spectrum of a single crystalline sample of 10 showing a sharp absorption at ṽOH=3522 cm−1. For comparison, an IR spectrum of crystalline phenol measured on the same IR spectrometer shows a ṽOH at 3223 cm−1 unambiguously indicating the presence of O−H⋅⋅⋅O hydrogen bond. Notably, instead of showing an O−H⋅⋅⋅O hydrogen bond, CycloSiFA 10 forms a centrosymmetric head-to-tail dimer via a weak H1⋅⋅⋅centre C11 A/C12 A interaction at a distance of 2.543(1) Å. Most remarkably, there is a O1⋅⋅⋅O1B interaction at a distance of 2.870(1) Å being shorter than twice the van der Waals radius29a of oxygen (1.52 Å). This situation resembles the unusual F⋅⋅⋅F interactions recently reported for {(F3C)3CO}3SiH which were interpreted as originating from London dispersion forces.29b
The next step was the preparation of alkynyl-substituted CycloSiFA 13 from the corresponding hydroxyl-substituted derivative 10 and propargyl bromide via nucleophilic substitution (Scheme 5). The desired propargyl ether was isolated in quantitative yield and fluorinated to afford SiFA compound 14. The alkyne 13 can easily be linked to other biomolecules to afford radiotracer precursors by click reaction.30 Copper- and ruthenium-catalysed azide-alkyne cycloadditions (CuAAC and RuAAC) are amongst the widely used click reactions that allow the formation of biologically stable 1,2,3-triazoles. Thus, as a test reaction, CuAAC between the alkyne derivative 13 and benzyl azide afforded the 1,4-disubstituted 1,2,3-triazole 15 in excellent yield of 95 %. Fluorination of the reaction product gave the SiFA-fluoride 16 in very good yield (94 %). Similar, RuAAC of 13 with benzyl azide afforded the 1,5-disubstituted 1,2,3-triazole 17 in excellent yield of 97 %. The substitution patterns of both cycloaddition products were determined using 1H−15N-HMBC NMR experiments. The successful realization of both click reaction conditions together with the stability of the CycloSiFAs should enable the formation of on-demand libraries of various radiotracers. Moreover, differently substituted CycloSiFAs could be synthesized for different applications, such as azides, maleimides, amines, aldehydes or tetrazines. To enhance the solubility of the cyclic derivatives in aqueous media for in vivo use, one could add water soluble linkers.20

Synthesis of the triazoles 15 and 17 by the reaction of the propargyl ether 13 with benzyl azide, BnN3, catalysed by tetraacetonitrile copper hexafluorophosphate, [Cu(MeCN)4]PF6, and pentamethylcyclopentadienyl bis(triphenylphosphane) ruthenium chloride, [Cp*(PPh3)2Ru]Cl, respectively (CuAAC- and RuAAC-type reactions).
To broaden the library of possible CycloSiFA compounds, the CycloSiFA derivative 24 bearing a hydroxymethyl substituent was prepared in a similar manner as the hydroxy-substituted compound 10 (Scheme 6).

Synthesis of CycloSiFA 24 containing a hydroxymethyl substituent and deprotection/ring opening.
Starting with the diol 18,31 a statistical TBS-protection of a single hydroxy group was performed to give the silyl ether 19 in 32 % yield.32 Oxidation of the unprotected alcohol with pyridinium chlorochromate (PCC) and subsequent condensation of the aldehyde with Dipp-amine afforded the imine 20 in 96 % yield over two steps. Halogen-lithium exchange and substitution with iPr2SiHCl gave the silane 21 in 73 % yield. After cyclisation to the TBS-protected CycloSiFA 22 in the presence of (C6F5)3B (91 % yield), the same reactivity pattern in deprotection/fluorination reactions was observed as for the silyl ether 9. With 2.0 molar equiv. of conc. hydrofluoric acid, complete cleavage of the Si−N-bond was observed after a reaction time of 15 min to give compound 23 in quantitative yield. With 6.0 molar equiv. of conc. hydrofluoric acid, additional cleavage of the silyl ether was achieved after 22 h reaction time to give the benzyl alcohol 25. Simple deprotection of the TBS-ether 22 with TBAF in THF proceeded much slower compared to phenol 9, yielding the desired CycloSiFA 24 in 88 % yield after a reaction time of 3 d at room temperature. In the presence of conc. HF, 24 undergoes facile ring opening to afford 25 (86 % yield). The overall yield for the synthesis of compound 24 starting from TBS-protected alcohol 19 was 56 % over five steps.
The first 18F-labelling approach of CycloSiFA revolved around our established “cold” non-radioactive fluorination protocol, employing aqueous 18F− and the labelling precursors 4 dissolved in CH2Cl2 to yield [18F]6. From previous experiments, the “cold” fluorinations of 4 resulting in the formation of 6 were performed in a non-mixable liquid-liquid two-phase system consistent of 4 dissolved in CH2Cl2 and the 18F− source provided as an aqueous solution of 18F− in 18O-enriched water (Scheme 7).

Upper row: Radiochemical fluorination of 4 to [18F]6 using ultrasound (100 μL CH2Cl2+40 μL H2O). Only 1–2 % of 18F- were transferred from the [18O]H2O phase into the CH2Cl2 phase. Thin layer chromatography (TLC) evaluation of the CH2Cl2 layer revealed a radiochemical conversion (RCC) of 18F− into [18F]6 of 79±16 % (based on only 1–2 % of 18F− being transferred to the CH2Cl2 phase). Bottom row: Radiofluorination of 4 and 13 in acetonitrile yielding [18F]6 and [18F]14 in RCC of 81±5 % and 90±5 %, respectively. The RCC in all cases was determined by radio-TLC.
The [18O]H2O is used in the cyclotron production of the 18F isotope where an 18O isotope is converted in a (p, n) nuclear reaction into an 18F isotope. The theoretical molar activity of 18F (Am, defined as the 18F radioactivity amount divided by the total molarity of 18F and 19F impurities) is 63 TBq/μmol and cannot be reached in practice.33-40 Non-radioactive 19F, inevitably diluting 18F, mostly stems from the tubing material used to move the 18F solution from the cyclotron target chamber to the automated synthesis unit for further processing. Consequently, the Am of 18F is found to much lower being in the range of 22–9900 GBq/μmol with one report referring to a measured Am of 43 TBq/μmol at the end of bombardment. In our labelling scenario we only used radioactivity amounts in the range of 300–500 MBq following the ALARA principle to reduce radiation exposure in our manual labelling setup.
Oxalic acid was added to the [18O]H2O phase before sonication to lower the pH of the [18O]H2O from 4.4 to 1.4 as it was found that no radiochemical conversion, hereafter referred to RCC, was observed at a pH of 4.4. To optimize 18F-fluorination using sonication, we had to establish the optimal depth of immersion of the reaction vial into the ultrasound bath to increase liquid-liquid phase interference for efficient radioactivity exchange between the two liquid phases. This process proved to be labour intensive and yielded only low 18F-transfer rates. The influence of using different volumes of 18F−/H2O and CH2Cl2 on the RCC yielding [18F]6 were investigated. Extensive evaluation revealed that even under optimized conditions (cf. experimental part), only 1–2 % of the 18F-radioactivity from the aqueous [18O]H2O phase was transported into the CH2Cl2 layer. Analysis of the CH2Cl2 radioactivity content using radio-thin layer chromatography (TLC) revealed that only 79±18 % of the radioactivity present in the CH2Cl2 phase accounted for [18F]6 (n=8). The rest radioactivity was identified by TLC being unreacted 18F–. Neither increasing the concentration of 4 nor adding a phase transfer catalyst such as K222 to the aqueous and CH2Cl2 phases improved the 18F-transfer rate or the RCC. The technical difficulties encountered (e.g. finding the optimal depth of immersion) and the low efficiency of 18F-transfer between the two phases rendered this labelling approach unsuitable for radiotracer development und automation.
In order to optimize 18F-labelling of 4 and to make the protocol more applicable to future automation, we developed a homogeneous one-phase liquid labelling method in acetonitrile. This methodology accounts for the unique conditions required for 18F-radiolabelling and prospective automation. The low amount of [18F]F– (M=pico- to nano-molar) used in our experiments (300–500 MBq were used) and short half-life of 18F (t1/2=110 min) necessitate the enhancement of 18F– anion nucleophilicity and the ability to quickly purify the labelling reaction mixture by solid phase extraction or high performance liquid chromatography (HPLC). The introduction of a homogeneous one-phase reaction system is a common denominator in most 18F-fluorination reactions based on nucleophilic [18F]F– anion labelling. Acetonitrile is an excellent dipolar aprotic solvent for 18F-radiofluorination and has proven its value in the development of the first-generation SiFA technology.
The [18F]F– was extracted from the aqueous solution obtained after the cyclotron bombardment of enriched [18O]water with protons, using a preconditioned potassium carbonate loaded quaternary methyl ammonium (hereafter referred to QMA) cartridge (46 mg K2CO3). The key to a successful CycloSiFA labelling in acetonitrile hinged on the sufficient acidification of the [18F]F– anion obtained from the QMA elution using tetraethyl ammonium tosylate (Et4NOTs) in acetonitrile containing a small amount of water.41 To efficiently elute the [18F]F– from the QMA, Et4NOTs (10 mg, 33 nmol) was dissolved in a mixture of 300 μL of water and 700 μL of acetonitrile. Previously, we disclosed the “four-drop method” where more than 90 % of the [18F]F– radioactivity was eluted from the QMA cartridge within the first four drops.21 Applied to our labelling scenario, this volume minimized eluate (ca. 150 μL) was azeotropically dried with acetonitrile to remove traces of water. An aliquot of the basic 18F-labelling cocktail was acidified using an optimized amount of oxalic acid and dissolved in acetonitrile to provide H[18F]F necessary for the CycloSiFA ring opening to proceed.
Two compounds were tested in this labelling scenario, compound 4 and the alkyne-derivatized precursor 13 (cf. Table 1, Scheme 5). The amount of oxalic acid had to be optimized for both reactions (cf. experimental part). Since the labelling reaction was performed at room temperature, the likelihood of H[18F]F evaporating from the reaction mixture was minimized. After a reaction time of 10 min at room temperature, an aliquot was taken and analysed using radio-TLC and radio-HPLC. Radiochemical conversion rates were found to be between 81±5 % and 90±5 % for 4 and 13, respectively (Scheme 7). [18F]14 was isolated for potential further prosthetic group Cu-catalysed click-radiolabelling using HPLC. The overall non-decay corrected radiochemical yield (RCY) was 46±4 % (n=3) after HPLC purification (Figure 1).

Crude reaction mixture of the formation of [18F]14. The aliquot that was injected onto HPLC was spiked with a small amount of non-radioactive 14 to confirm the identity of the radioactive HPLC peak (blue line=radioactivity channel; red line=UV channel).
Comparing the TLC results with the HPLC preparative yields revealed that some of the HPLC observed side products could not be identified via TLC. This explains the discrepancy between the RCC obtained from TLC readings and the preparative HPLC RCYs. Finally, the [18F]14 was separated from the HPLC solvent by solid phase cartridge extraction. [18F]14 and other CycloSiFA compounds currently in development will be used in future radiolabelling reactions.
Conclusion
We have synthesized a variety of novel azasilole derivatives (five-membered rings) and proved their ability to undergo ring-opening by fluoride anion under mild and efficient conditions (low temperature, fast reaction, no by-product separation needed). Some of the azasiloles contain at the aryl backbone functional groups which are able to undergo click-type chemistry allowing straightforward linkage to biomolecules on demand. This opens a new chapter of silicon-based 18F radiochemistry for PET tracer development fueled by SiFA's already perceptible clinical success. PET technology has been forecasted to see an enormous growth within the next decade. The concept holds potential to be extended to six-membered rings (for which ring-opening could proceed even faster and milder) and also to boron-based analogues. Especially tumour homing peptides (e.g. octreotide and octreotate) for diagnostic oncologic PET imaging could provide potential targets for CycloSiFA radiolabelling. We will further investigate whether our labelling conditions lead to a premature hydrolysis of the CycloSiFA precursor alongside the 18F-radiolabelling. By adding a silanol by-product to the reaction mixture, the effective Am of the 18F-SiFA compound could be negatively affected. Obtaining higher molar activities in an optimized CycloSiFA labelling setup could provide an advantage over the currently used SiFA-based radiopharmaceuticals and consolidate its value as a new entry into 18F-labelling technologies.
Acknowledgments
This work has been supported by the Natural Sciences and Engineering Research Council of Canada (NSERC), Discovery Grant to RS. The authors M.M., J.B., L.I., N.K., and K.J. are grateful to TU Dortmund for supporting this work. We thank the anonymous reviewers for valuable hints. Open Access funding enabled and organized by Projekt DEAL.
Conflict of interest
The authors declare no conflict of interest.
Open Research
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.

