

B-Cu-Zn Gas Diffusion Electrodes for CO2 Electroreduction to C2+ Products at High Current Densities
Graphical Abstract
A B-Cu gas diffusion electrode (GDE) system was developed for highly selective CO2 conversion to C2+ products at high relevant current densities by mitigating the flooding problem and tuning the three-phase boundary. Anodic protection by sacrificial Zn nanosheets remarkably improved the long-term stability for CO2 electroreduction at high current densities.

Abstract
Electroreduction of CO2 to multi-carbon products has attracted considerable attention as it provides an avenue to high-density renewable energy storage. However, the selectivity and stability under high current densities are rarely reported. Herein, B-doped Cu (B-Cu) and B-Cu-Zn gas diffusion electrodes (GDE) were developed for highly selective and stable CO2 conversion to C2+ products at industrially relevant current densities. The B-Cu GDE exhibited a high Faradaic efficiency of 79 % for C2+ products formation at a current density of −200 mA cm−2 and a potential of −0.45 V vs. RHE. The long-term stability for C2+ formation was substantially improved by incorporating an optimal amount of Zn. Operando Raman spectra confirm the retained Cu+ species under CO2 reduction conditions and the lower overpotential for *OCO formation upon incorporation of Zn, which lead to the excellent conversion of CO2 to C2+ products on B-Cu-Zn GDEs.
Introduction
Worldwide climate change has become a regular topic in the news, as anthropogenic CO2 levels have continued to steadily increase.1-3 The direct conversion of CO2 into chemical feedstocks or high energy-density fuels driven by renewable electricity is a potentially attractive technology to negate CO2 emissions. However, despite many efforts being devoted to improving electrochemical CO2 valorisation, development is still in the early-stages due to sub-optimal activity and product selectivity.4-6 Employing gas diffusion electrode (GDE) systems has emerged as a promising approach to promote the electrochemical CO2 reduction reaction (eCO2RR) towards possible applications.3, 7 By overcoming the low solubility of CO2 in aqueous electrolytes (approx. 35 mm) and shortening the diffusion path for the reaction, GDE-based systems can potentially reach industry-relevant current densities as opposed to the bathed electrodes utilized in a conventional H-cell.7-9
As catalyst for eCO2RR, Cu has been intensively studied due to its unique ability to produce hydrocarbons and alcohols, which is attributed to a moderate CO* binding energy.10, 11 The pre-oxidation of Cu has previously been reported to greatly improve the intrinsic catalytic properties towards C2+ products formation.12 The catalytic role played by Cu+ and subsurface oxygen was later revealed, where Cu+ interacts synergistically with Cu0 to promote C2+ products formation owing to the lower energy barrier for C−C coupling.13-18 Despite some recent efforts on plasma-activated Cu,16 electro-redeposited Cu,17 and multi-hollow Cu2O,18 the stabilisation issue of Cu+ under eCO2RR condition remains challenging.
Introducing modifier elements has been considered as an efficient strategy to reduce the tendency of Cu+ reduction at negative potentials.19-22 Boron has an electron configuration of 2s22p1 and thus exhibits Lewis acidity. The empty electronic orbital of B tends to receive electrons from Cu, tuning the local electronic structure of Cu with positive valence sites and making it more Lewis acidic, both of which are favourable for boosting the formation of C2+ products in eCO2RR. A B-doped oxide-derived-Cu catalyst exhibited a higher Faradaic efficiency (FE) of 48.2 % toward the formation of C2+ products than that of its boron-free oxide-derived-Cu counterpart (30.5 %), owing to stabilisation of the Cu+ species by the introduction of B.21 By doping different amounts of B, the valence state of Cu could be tuned to achieve optimum C2+ products formation with a high FE of 79 % at −1.1 V vs. RHE in 0.1 m KCl.22 However, the current densities in these reports (≤70 mA cm−2) are far below any possible application.
In addition to a high FE towards particular desirable products and high current density, stable long-term operation is also highly important for viable application of a given catalyst for eCO2RR.23, 24 In addition to the challenge of inherent chemical instability of Cu-based catalysts, catalyst degradation and cathodic corrosion under high cathodic potentials are similarly severe drawbacks.7, 23-27 Inhibition of these processes to stabilize Cu-based catalysts is important yet still challenging for the development of efficient eCO2RR systems.
Herein, we report a B-doped Cu (B-Cu) catalyst for efficient eCO2RR at industry-relevant current densities using a GDE electrolysis system. During operation especially at high current densities, GDEs are suffering from flooding due to electrowetting. Notably, this flooding problem, which characteristically leads to a low concentration of CO2 at the triple-phase boundary due to low CO2 solubility, was mitigated by controlling the amount of the hydrophobic binder PTFE (polytetrafluoroethylene) during catalyst ink preparation and the gas-liquid-catalyst interphase was further optimized by tuning the catalyst loading. As a result, a highest FE of 78 % for C2+ products formation (C2H4 49 %, C2H5OH 22 %, C3H7OH 7 %) was achieved on B-Cu GDE with 10 % of PTFE and 0.5 mg cm−2 catalyst loading at −200 mA cm−2 and −0.45 V vs. RHE. Furthermore, a sacrificial anode of Zn, a less noble metal, was employed to stabilize B-Cu catalyst during eCO2RR. With an optimum amount of incorporated Zn nanosheets (0.025 mg cm−2), the long-term stability for C2+ product formation at −200 mA cm−2 was significantly improved with an overall C2+ product current density of −194 mA cm−2. Operando electrochemical Raman spectroscopy (OERS) results show that the active Cu+ species is stable even at high reduction potentials, the overpotential for *OCO formation is positively shifted by incorporation of Zn nanosheets, both of which contribute to the excellent eCO2RR performance of B-Cu-Zn GDE.
Results and Discussion
The B-Cu catalyst was synthesized by a facile and scalable method22, 28, 29 using concentrated sodium borohydride (NaBH4) as both the reducing agent and boron source under continuous Ar purging. Zn nanosheets were synthesized by electrochemical deposition of Zn on a Cu foil followed by scratching them off. Figure 1 A displays the X-ray diffraction (XRD) pattern of the as-synthesized B-Cu sample. The main peaks at 2θ values of 43.3°, 50.4°, and 74.1° are corresponding to the (111), (200) and (220) planes, respectively, of cubic metallic Cu with a space group of Fm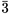 m (COD: 9013014). In addition, weak peaks of cubic Cu2O (111), (200), and (220) planes with a space group of Pn
m (COD: 9013014). In addition, weak peaks of cubic Cu2O (111), (200), and (220) planes with a space group of Pn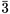 m (COD: 1000063) are observed due to oxidation in air or Cu-B interaction with the solvent during synthesis. Similarly, peaks assigned to hexagonal Zn (P63/mmc, COD: 9012435) and ZnO (P63mc, COD: 9008877) are both observed in the XRD pattern of the as-synthesized Zn sample (Figure 1 B), with the intensity of the oxide peaks being notably higher compared to B-Cu due to faster oxidation of metallic Zn.
m (COD: 1000063) are observed due to oxidation in air or Cu-B interaction with the solvent during synthesis. Similarly, peaks assigned to hexagonal Zn (P63/mmc, COD: 9012435) and ZnO (P63mc, COD: 9008877) are both observed in the XRD pattern of the as-synthesized Zn sample (Figure 1 B), with the intensity of the oxide peaks being notably higher compared to B-Cu due to faster oxidation of metallic Zn.

XRD patterns of A) B-Cu nanoparticles and B) Zn nanosheets. TEM images of C) B-Cu nanoparticles and D) Zn nanosheets. HRTEM images of E) B-Cu nanoparticles and F) Zn nanosheets.
The transmission electron microscopy (TEM) image (Figure 1 C) reveals the nanoparticle morphology of the as-synthesized B-Cu sample with some degree of agglomeration. The corresponding size distribution histogram (Figure S1) shows that the B-Cu nanoparticle sizes were in the 23–31 nm range. The as-synthesized Zn sample (Figure 1 D) exhibits nanosheet morphology. High-resolution transmission electron microscopy (HRTEM) images of the B-Cu sample (Figure 1 E) show lattice fringes with three different d-spacings of 3.01, 2.46, and 2.13 Å, which correspond to Cu2O (110), (111), and (200), respectively, while no metallic Cu-related lattice fringes were observed. This is most likely due to the surface oxide-covered metallic Cu phase making it difficult to be observed by HRTEM. Similarly, only ZnO (100) and (101) lattice fringes with d-spacings of 2.81 and 2.47 Å, respectively, were identified in Zn nanosheets (Figure 1 F).
X-ray photoelectron spectroscopy (XPS) was employed in order to study the chemical states and composition of the catalysts. The XP spectrum of B-Cu nanoparticles presents a sharp Cu 2p3/2 peak at 932.4 eV (Figure S2), which can be assigned to Cu0 and/or Cu+ species.30 To distinguish Cu0 and Cu+, the Cu LMM Auger transition (Figure 2 A) was additionally recorded, which indicates that the surface of B-Cu nanoparticles mainly consisted of a Cu+ (Cu2O) oxidation state at a kinetic energy of 916.7 eV. This result is consistent with the HRTEM analysis. The presence of B in B-Cu was confirmed by B 1s XPS. Only B sub-oxide signals at about 191.0 eV were observed in the B 1s spectrum (Figure 2 B), since the solubility of B in Cu is extremely low and elemental B is typically oxidized at the surface. The B:Cu atom ratio in the B-doped Cu sample is 0.0144 as determined by XPS. Figure 2 C presents the Zn 2p XP spectrum of Zn nanosheets with a sharp Zn 2p3/2 peak at 1022 eV. Zn0 and Zn2+ (ZnO) can be distinguished using the Zn LMM spectrum (Figure 2 D), in which the well-resolved peak at around 987.8 eV is assigned to ZnO. ZnO as the main component on the surface of Zn nanosheets matches well with the HRTEM analysis in Figure 1 F.

A) Cu LMM and B) B 1s XPS spectra for B-Cu nanoparticles. C) Zn 2p and D) Zn LMM spectra for Zn nanosheets.
The electrocatalytic activity and selectivity of the catalysts towards eCO2RR were evaluated in a H-type GDE cell (Figure S3) employing 1 m aqueous KOH as electrolyte. In our experiments, the catalyst ink was directly drop-coated on carbon paper with a hydrophobic microporous gas diffusion layer (GDL) to obtain the GDE (see details in Supporting Information). GDEs having only B-Cu in the catalyst layer show that the predominant product during eCO2RR is H2 over the whole applied current range (Figure S4A). The Faradaic efficiency (FE) for H2 was 78 %, the FE for C1 products (CO and formate) was 4.8 %, and the FE for C2 products (C2H4 and C2H5OH) was 5.8 %, at a current density of −200 mA cm−2 and a potential of −0.6 V vs. RHE (Figure 3 A, Figure S4A). The comparatively high FE for H2 evolution can be ascribed to the high hydrophilicity of the B-Cu catalyst as demonstrated by contact angle measurements (Figure S5). Moreover, the hydrophilic property of B-Cu accelerates flooding of the GDE, thus limiting eCO2RR. At a current density of −400 mA cm−2, H2 is the only detectable product, and the electrolyte penetrated the whole GDE as leakage was observed.

A) FE of H2, C1, and C2+ products on B-Cu electrodes with 0, 5, 10, and 20 % PTFE at a current density of −200 mA cm−2 and a potential of −0.60, −0.59, −0.54, and −0.54 V vs. RHE, respectively. B) C2+/C1 ratio and C) partial current density of C2+ products at different potentials on B-Cu electrodes with different contents of PTFE. D) FE for the formation of H2, C1, and C2+ products on B-Cu electrodes with 0.1, 0.2, 0.5, and 1.0 mg cm−2 at a current density of −200 mA cm−2 and a potential of −0.64, −0.53, −0.45, and −0.54 V vs. RHE, respectively. E) FE for the formation of various C2+ products at a catalyst loading of 0.5 mg cm−2 B-Cu at different potentials. F) Partial current density of C2+ products at different potentials on B-Cu electrodes with different loadings. Product quantification was performed by means of gas chromatography (Figure S6) and 1H-NMR spectroscopy (Figure S7).
To mitigate the flooding problem, we added varying amounts of PTFE to the catalyst ink to enhance the hydrophobicity of the B-Cu catalyst layer on top of the GDE. By increasing the content of PTFE relative to the total mass loading of the catalyst from 5 to 10 and 20 %, the hydrophobicity of the B-Cu GDE gradually increased as shown in the corresponding contact angle measurements (Figure S5). As expected, the hydrogen evolution reaction (HER) was substantially decreased and the formation of C2+ products, mainly ethylene and ethanol, was enhanced accordingly (Figure S4). For B-Cu-modified GDEs with 5 % PTFE, the FE for H2 decreased to 69 % at a current density of −200 mA cm−2 and a potential of −0.59 V vs. RHE, the FE for C1 products increased to 8.4 % (CO 3.1 %, formate 4.4 %), and the FE for C2+ products increased to 17 % (C2H4 5 %, C2H5OH 12 %; Figure 3 A, Figure S4B). When the content of PTFE was further increased to 10 %, the FE for H2 was further reduced to 28 % at a current density of −200 mA cm−2 and a potential of −0.54 V vs. RHE, at which the FE for C1 products decreased to 5 % (CO 4 %, formate 1.0 %), and the FE for C2+ products increased to 70 % (C2H4 43 %, C2H5OH 23 %, C3H7OH 4 %; Figure 3 A, Figure S4C). However, further increase in the PTFE content to 20 % did not lead to a decrease in the HER (32 % at −200 mA cm−2, −0.54 V vs. RHE). Concomitantly, the FE for C1 products increased to 8 % (CO 6 %, formate 2 %), and the FE for C2+ products decreased to 53 % (C2H4 27 %, C2H5OH 20 %, C3H7OH 5 %; Figure 3 A, Figure S4D). This is most likely due to a decrease in conductivity of the B-Cu catalyst/PTFE layer and possible blocking of the active sites of the catalyst. B-Cu GDEs with 10 % PTFE exhibited the maximum FE for the formation of C2+ products and the minimum FE for H2 and C1 products formation.
The selectivity of eCO2RR to C2+ products on B-Cu GDEs with different contents of PTFE can be further evaluated by the FE for the C2+/C1 products ratio. As shown in Figure 3 B, B-Cu-10 %PTFE is highly selective for the formation of C2+ products at potentials more negative than −0.43 V vs. RHE. Particularly, at −0.59 V vs. RHE and −300 mA cm−2, the C2+/C1 ratio of B-Cu-10 %PTFE reaches 14.4, which is superior to electrodes with 0, 5, and 20 % PTFE. The selectivity of eCO2RR to C2+ products on B-Cu-10 %PTFE GDEs reached up to 93.5 % (Figure S8).
The overpotential for eCO2RR at a total current density of −10 mA cm−2 decreases with increasing PTFE added to the B-Cu GDE (Figure S9). The total current density, as well as the partial current densities for C2+ product formation were greatly enhanced upon the addition of PTFE on the B-Cu GDE (Figure 3 C). B-Cu-10 %PTFE shows the maximum C2+ geometric current density of −184 mA cm−2 at −0.59 V vs. RHE, which is around 16 times higher than that obtained using B-Cu without PTFE (−11.6 mA cm−2). We conclude that the intermediate amount of PTFE (10 %) can balance hydrophobicity and conductivity of B-Cu and mitigate the flooding problem of the GDE, thereby boosting the conversion of CO2 towards C2+ compounds.
The three-phase interphase within the GDE plays a key role for the eCO2RR, which can be affected by the thickness of the catalyst layer. Hence, we further changed the mass loading of the catalyst to investigate the effect of catalyst thickness on the eCO2RR at the three-phase interphase. The thickness of the catalyst layer on the top of GDL gradually decreases from 26.7 μm to 13.4, 5.5 and 2.5 μm when decreasing the mass loading of the catalyst from 1 mg cm−2 to 0.5, 0.2 and 0.1 mg cm−2, respectively (Figure S10). With decreasing mass loading of B-Cu, the FE for H2 formation was further decreased to about 20 % with a catalyst loading of 0.5 and 0.2 mg cm−2, respectively, at −200 mA cm−2, while it increased to 56 % when only 0.1 mg cm−2 of the catalyst was used (Figure S11). In the case of a too thin catalyst layer, the electrolyte will penetrate into the GDL causing flooding which is ultimately promoting the HER and inhibiting the eCO2RR. For example, on B-Cu GDEs with a mass loading of only 0.1 mg cm−2, H2 formation accounted for 85 % of the current at −300 mA cm−2. Further increase of the current density to −400 mA cm−2 led to increasing electrowetting and hence penetration of the electrolyte through the GDE, ultimately causing the GDE to lose its function.
The formation of CH4 was completely suppressed at a loading of 1.0 mg cm−2, however, decreasing the loading to 0.5 mg cm−2 led to CH4 formation with a FE of around 0.02 % at −200 mA cm−2. The FE for CH4 formation gradually increased to 0.5 and 8 % at loadings of 0.2 and 0.1 mg cm−2, respectively (Figure S11, S12), suggesting that the formation of C1 products increases with decreasing catalyst loading (Figure 3 D). Typically, at −200 mA cm−2, the FE for C2H4 first increased to 49 % for loadings of 0.5 and 0.2 mg cm−2, and then decreased to 18 % for a loading of 0.1 mg cm−2, whereas the FE for C2H5OH gradually decreased to 22 and 18 % for loadings of 0.5 and 0.2 mg cm−2, and further decreased at 14 % for a loading of 0.1 mg cm−2 (Figure S11). In summary, the FE for C2+ compounds including C2H4, C2H5OH, C3H7OH, and acetate was 78 % for a loading of 0.5 mg cm−2 (Figure 3 D), consisting of 49 % C2H4, 22 % C2H5OH, 7 % C3H7OH at a current density of −200 mA cm−2 and a potential of −0.45 V vs. RHE (Figure 3 E).
The current density at potentials more negative than −0.40 V vs. RHE first increases by decreasing the catalyst loading from 1 to 0.5 mg cm−2 and then decreases with further lowering the mass loadings to 0.2 and 0.1 mg cm−2 (Figure S13), which coincides with the partial current for the eCO2RR. This can be further confirmed by a comparison of the partial current densities for C2+ formation as shown in Figure 3 F. The highest C2+ current density of −198 mA cm−2 (at −0.49 V vs. RHE) was achieved at a loading of 0.5 mg cm−2 of B-Cu GDE. In contrast, the maximum C2+ current density for catalyst loadings of 1.0, 0.2, and 0.1 mg cm−2, was 184, 152, and 69 mA cm−2 at more negative potentials of −0.59, −0.53, and −0.56 V vs. RHE, respectively. This implies that the mass loading of the B-Cu catalyst, and thus the thickness of the catalyst layer, considerably influences the location of the three-phase interphase and the contribution of eCO2RR to the overall current. A suitable penetration depth of the electrolyte on the one hand together with a sufficient diffusion depth of gaseous CO2 on the other hand are necessary to maximize the activity and efficiency of eCO2RR.
To address the cathodic corrosion issue of Cu catalysts at high cathodic potentials, which limits the long-term stability of such catalysts, the less noble metal Zn was employed as sacrificial anode to protect the B-Cu catalyst. For this, a B-Cu-Zn catalyst was prepared by physical mixing of B-Cu nanoparticles and Zn nanosheets on a carbon paper as support. The B-Cu loading was fixed at 0.5 mg cm−2 while the Zn loading was increased gradually from 0.01 to 0.075 mg cm−2. The maximum FEs for the formed C2H4, C2H5OH and C3H7OH and their corresponding potentials can both be adjusted by varying the Zn loading in the B-Cu-Zn catalyst (Figure S14). The catalyst with a composition of 0.5 B-Cu:0.025 Zn showed the highest FE for alcohols (34 %), with C2H5OH (31 %) and C3H7OH (3 %) at −0.45 V vs. RHE and a total current density of −200 mA cm−2 (Figure 4 A,B). Further increasing the Zn loading results in a decline of the maximum FE for the formation of C2H5OH, which can be attributed to the excess CO generated on Zn (Figure S15) and the competing HER. The maximum FE for the formation of C2H4 monotonically decreases from 49 % on B-Cu to 27 % on 0.5 B-Cu:0.075 Zn. Hence, the selectivity for alcohols versus C2H4 (FE alcohols / FE C2H4) can be tuned from 0.57 on a bare B-Cu electrode to 1.04 on a 0.5 B-Cu:0.075 Zn electrode by the amount of added Zn nanosheets (Figure 4 A).

A) FE for the formation of C2H4 and alcohols and the ratios of alcohols/C2H4 on B-Cu electrodes with different amounts of Zn at a current density of −200 mA cm−2. B) FE for the formation of various C2+ products on 0.5 B-Cu:0.025 Zn electrodes at different potentials. C) Partial current density for the formation of C2+ products at different potentials on B-Cu electrodes with different amounts of Zn. D) FE for the formation of various C2+ products on 0.5 B-Cu and 0.5 B-Cu:0.025 Zn GDEs at a current density of −200 mA cm−2 and 4 h of electrolysis.
The current density at potentials more positive than −0.45 V vs. RHE was highest using a 0.5 B-Cu:0.025 Zn GDE, while at potentials more negative than −0.45 V vs. RHE, the current density was highest at a higher Zn loading of 0.5 B-Cu:0.05 Zn GDE (Figure S16). Further increasing the Zn loading to 0.075 mg cm−2 resulted in a drop of current density due to the excess of Zn and fewer Cu active sites for eCO2RR. The partial current densities for the formation of C2+ products are further compared in Figure 4 C. Using a 0.5 B-Cu:0.025 Zn GDE, the highest current density for the formation of C2+ products is obtained over almost the whole potential range, with an observed value of −194 mA cm−2 at −0.49 V vs. RHE being similar to that of the B-Cu electrode. In contrast, using GDEs with catalyst loadings of 0.5 mg cm−2 B-Cu GDEs mixed with 0.01, 0.05, and 0.075 mg cm−2 Zn nanosheets, maximum C2+ product formation current densities of −158, −156, and −120 mA cm−2 were obtained at potentials of −0.52, −0.47, and −0.56 V vs. RHE, respectively. Hence, an optimum amount of Zn (0.025 mg cm−2) is required for minimizing cathodic Cu corrosion while retaining reasonable activity of C2+ product formation.
The long-term eCO2RR stability of the catalyst is one of the key factors for potential applications, notwithstanding the fact that maintaining the catalyst integrity still remains challenging for GDE-based systems. The stability of the 0.5 B-Cu:0.025 Zn electrode in comparison with a B-Cu electrode was evaluated by performing eCO2RR electrolysis at a high current density of −200 mA cm−2. The experimental results shown in Figure 4 D and Figure S17 reveal excellent stability of the 0.5 B-Cu:0.025 Zn electrode with a negligible decay of the FE for the formation of C2+ products. In contrast, the FE for the formation of C2+ products on B-Cu GDEs starts to attenuate after 1 h of eCO2RR electrolysis at the expense of increasing H2 formation. Thus, the incorporation of Zn nanosheets remarkably improved the long-term eCO2RR stability of the catalyst, however with sacrificing a little bit of the FE for the formation of C2+ products at the beginning of the electrolysis. To better understand the difference in the observed long-term stability of these two kinds of GDEs, the change in composition and chemical states after electrocatalysis was further investigated by recording Cu and Zn LMM Auger spectra before and after the stability test. Qualitative assessment of the Cu LMM Auger spectra (Figure S18) reveals Cu+ with a peak maximum at around 916.3 eV kinetic energy to be the main surface component in both the 0.5B-Cu:0.025Zn GDE and the B-Cu GDE even after long-term eCO2RR. Besides Cu+, a contribution of metallic Cu manifests as a shoulder at a kinetic energy of around 918.5 eV. Interestingly, that contribution diminishes nearly completely after 4 h of electrolysis for the B-Cu GDE, whereas the 0.5B-Cu:0.025Zn GDE retains a visible shoulder corresponding to Cu0 after the same duration of electrolysis. These findings demonstrate the stabilization of the metallic Cu state on the GDE surface by Zn in 0.5B-Cu:0.025Zn. Further evidence for the sacrificial anodic protection effect of Zn is obtained from the Zn LMM Auger spectra on 0.5B-Cu:0.025Zn GDE before and after 15 min of eCO2RR (Figure S19). While the as-prepared GDE shows similar contributions of metallic Zn and ZnO, the GDE after 15 min of eCO2RR exhibits a stronger decay of the Zn0 peak intensity compared to the one of ZnO. Notably, no traces of surface Zn are detectable after 4 h of electrolysis (spectrum not shown) confirming the anodic Zn corrosion process concomitantly stabilizing Cu+ and Cu0. This finding does not only explain the higher stability of the 0.5B-Cu:0.025Zn GDE, but also confirms the synergistic function of the presence of Cu+ and Cu0 to promote C2+ product formation. The obtained Cu LMM Auger spectra (Figure S18) confirmed that both the 0.5 B-Cu:0.025 Zn GDE and the B-Cu GDE mainly exhibit Cu+ on the surface even after long-term eCO2RR. Besides Cu+, a fraction of Cu0 was also observed which marginally increased during electrolysis. The 0.5 B-Cu:0.025 Zn GDE shows a more stable Cu+/Cu0 ratio after 4 h of electrolysis with a lower Cu+/Cu0 ratio due to the Zn protection as compared with the 0.5 B-Cu GDE. This finding does not only explain the higher stability of the 0.5 B-Cu:0.025 Zn GDE, but also confirms the synergistic function of the presence of Cu+ and Cu0 to promote C2+ product formation. The obtained Zn LMM Auger spectra on 0.5 B-Cu:0.025 Zn GDE (Figure S19) shows a significant decrease of both Zn content and metallic Zn after long-term eCO2RR, which confirms the sacrificial protection effect of Zn on the B-Cu catalyst.
To further investigate the origin of the eCO2RR performance of both the B-Cu and 0.5 B-Cu:0.025 Zn GDEs, operando Raman spectroscopy measurements were carried out by sequential application of potentials from 0.27 to −1.43 V vs. RHE. First, the structural evolution of the catalyst was monitored (Figure 5). Both 0.5 B-Cu and 0.5 B-Cu:0.025 Zn GDEs exhibit Raman peaks at 218, 522, and 640 cm−1, which were ascribed to the Cu2O phase.18, 31, 32 These Raman signals remain essentially unchanged under operando conditions demonstrating the stability of the active Cu+ species even at high reduction potentials. This is expected to contribute to the efficient conversion of CO2 to C2+ products at the comparatively high applied current densities. Operando electrochemistry Raman measurements were additionally used to detect possible intermediates of the eCO2RR on 0.5 B-Cu and 0.5 B-Cu:0.025 Zn electrodes. The peak at 1072 cm−1 is assigned to the anti-symmetrical stretching of *OCO,32, 33 which can be observed from a potential of below −0.23 V vs. RHE and increases in intensity at more cathodic potentials for the 0.5 B-Cu electrode (Figure 5 A). In contrast, the 0.5 B-Cu:0.025 Zn electrode already shows a sharp *OCO vibrational peak at a potential of 0.27 V vs. RHE, which gradually weakens at more cathodic potentials (Figure 5 B). This implies that the activation of CO2 can be achieved at more positive potentials for the 0.5 B-Cu:0.025 Zn electrode additionally supporting the observed higher eCO2RR activity for the 0.5 B-Cu:0.025 Zn electrode as compared with the 0.5 B-Cu electrode (Figure 4 C). Raman peaks at 283, 344, and 2095 cm−1 for both 0.5B-Cu and 0.5 B-Cu:0.025 Zn electrodes correspond to adsorbed CO on Cu,31-34 which represents the key intermediate for C−C coupling and C2+ products formation. The peak at about 2713 cm−1 is associated to the C−H vibration35 and further confirms the formation of hydrocarbons.

Operando electrochemistry Raman spectra for a A) 0.5 B-Cu GDE and a B) 0.5 B-Cu:0.025 Zn GDE during eCO2RR at different applied potentials.
Conclusion
A B-Cu GDE system was developed for selective CO2 conversion to C2+ products at seldom reported high current densities. The flooding problem in this GDE system, that is, liquid electrolyte domination at the triple-phase boundary, was mitigated through controlling the amount of added PTFE and by optimizing the catalyst loading leading to optimisation of the gas-liquid-catalyst interphase. The FE for the formation of C2+ products was as high as 78 % on a 0.5 mg cm−2 B-Cu GDE with 10 % PTFE at a current density of −200 mA cm−2 and a potential of −0.45 V vs. RHE. By incorporation of an optimum amount of Zn nanosheets (0.025 mg cm−2), the activity and long-term stability of the B-Cu catalyst were substantially improved reaching a maximum current density for the formation of C2+ products of −194 mA cm−2. Operando electrochemical Raman results demonstrate the stable presence of Cu+ species during eCO2RR and the positive shift of the overpotential for *OCO formation upon incorporation of Zn, which concertedly result in the excellent eCO2RR performance of the B-Cu-Zn GDE. These findings suggest a strategy for optimizing GDE structures to ultimately improve the eCO2RR activity, selectivity, and stability for the formation of value-added chemicals/fuels at high current densities which may become suitable for potential applications.
Acknowledgements
The project has received funding from the ERC (CasCat [833408]) and by the Deutsche Forschungsgemeinschaft in the framework of the research unit FOR 2397e2 (276655237) as well as under Germany's Excellence Strategy—EXC 2033–390677874—RESOLV. CA acknowledges funding by the BMBF in the framework of the NanomatFutur project “MatGasDif” (03XP0263). NS acknowledges the Alexander von Humboldt foundation for a Postdoc fellowship. Y.S. is grateful to the International Clean Energy Talent Program (iCET) hosted by the Future Energy Profile at Mälardalen University in cooperation with the China Scholarship Council (CSC) and the Applied Energy Journal. The authors acknowledge the support of Dr Ulrich Hagemann from the DFG core facility ICAN (DFG RI 00313) for a part of the XPS measurements. Sandra Schmidt is acknowledged for help with the SEM measurements. Open access funding enabled and organized by Projekt DEAL.
Conflict of interest
The authors declare no conflict of interest.

