

Generation and Stabilization of a Dinickel Catalyst in a Metal-Organic Framework for Selective Hydrogenation Reactions
Abstract
Although many monometallic active sites have been installed in metal–organic frameworks (MOFs) for catalytic reactions, there are no effective strategies to generate bimetallic catalysts in MOFs. Here we report the synthesis of a robust, efficient, and reusable MOF catalyst, MOF-NiH, by adaptively generating and stabilizing dinickel active sites using the bipyridine groups in MOF-253 with the formula of Al(OH)(2,2′-bipyridine-5,5′-dicarboxylate) for Z-selective semihydrogenation of alkynes and selective hydrogenation of C=C bonds in α,β-unsaturated aldehydes and ketones. Spectroscopic studies established the dinickel complex (bpy⋅−)NiII(μ2-H)2NiII(bpy⋅−) as the active catalyst. MOF-NiH efficiently catalyzed selective hydrogenation reactions with turnover numbers of up to 192 and could be used in five cycles of hydrogenation reactions without catalyst leaching or significant decrease of catalytic activities. The present work uncovers a synthetic strategy toward solution-inaccessible Earth-abundant bimetallic MOF catalysts for sustainable catalysis.
Introduction
Hydrogenation of unsaturated bonds is one of the most important chemical transformations for the production of commodity and fine chemicals due to the high atom economy and readily available starting materials.1 High selectivities, including chemo-,2 regio-,3 and stereoselectivity,4 are essential for fine chemical production and organic synthesis.5 Selective hydrogenation reactions are typically catalyzed by precious metal catalysts. For example, the Lindlar catalyst, a poisoned palladium catalyst on calcium carbonate or barium sulfate, has been used for semihydrogenation of alkynes to (Z)-alkenes.6 However, due to the low Earth-abundance, high costs, and inherent toxicity of precious metals, it is imperative to develop Earth-abundant metal catalysts for selective hydrogenation reactions.
As a unique class of porous molecular materials, metal–organic frameworks (MOFs),7 have been explored for a wide range of applications, including gas storage8 and separation,9 sensing,10 and biomedical imaging and cancer therapy.10, 11 MOFs are particularly suited for designing reusable porous single-site solid catalysts by taking advantage of their molecular tunability, active site access via large channels, and enhanced catalyst stability.12 Thus, MOF catalysts can combine the molecular tunability and uniform catalytic sites of homogeneous catalysts and the stability, facile separation, and reusability of heterogeneous catalysts to afford a new class of sustainable catalysts for organic transformations.13 In some examples, MOFs allow the stabilization of catalytically active centers via site isolation to design solution-inaccessible catalytic species based on single metal centers.14
Herein, we report the design and stabilization of novel dinickel active sites in an aluminum MOF for selective hydrogenation of unsaturated compounds (Figure 1). The active catalytic site in MOF-NiH was characterized by multiple spectroscopic methods as a bipyridine (bpy) linker-supported NiII2(μ2-H)2 species. MOF-NiH catalyzed semihydrogenation of alkynes to (Z)-alkenes and selective hydrogenation of C=C bonds in α,β-unsaturated aldehydes and ketones. The reaction mechanism was investigated and discussed.

Illustration of selective hydrogenation of alkynes as well as α,β-unsaturated aldehydes and ketones by the MOF-NiH catalyst with NiII2(μ2-H)2 active sites.
Results and Discussion
MOF-253,15 a DUT-5-type16 MOF with 2,2′-bipyridine-5,5′-dicarboxylate (dcbpy) linkers, was synthesized by heating a mixture of Al(NO3)3 ⋅ 9H2O, 2,2′-bipyridine-5,5′-dicarboxylic acid (H2dcbpy), acetic acid (HOAc), and N,N′-dimethylformamide (DMF) at 120 °C (Figure 2a). Powder X-ray diffraction (PXRD) studies showed that as-synthesized MOF-253 was highly crystalline with peak positions matching well with those of the literature report (Figure 2b).15, 17 Transmission electron microscopy (TEM) revealed a needle-like morphology for MOF-253 (Figure S2) whereas N2 sorption measurements showed type I isotherms with a Brunauer–Emmett–Teller (BET) surface area of 1466 m2/g, which is comparable with the literature value (Figure 2c).18

(a) Synthesis of MOF-253 and its metalation with NiBr2 followed by treatment with NaBHEt3 to afford MOF-NiH. (b) The similarity of PXRD patterns of MOF-253 (red), MOF-NiBr2 (blue), MOF-NiH (purple), and the recovered MOF-NiH (green) to the simulated pattern of MOF-253 (black). (c) N2 adsorption isotherms of MOF-253 (black) and MOF-NiBr2 (red) at 77 K. (d) TEM image of MOF-NiBr2.
The as-synthesized MOF-253 was metalated with NiBr2(1,2-dimethoxyethane) [NiBr2(dme)] in acetonitrile to afford the precatalyst MOF-NiBr2 (Figure 2a). Inductively coupled plasma-mass spectroscopy (ICP-MS) of digested MOF-NiBr2 showed a Ni : Al ratio of 1.00±0.02, indicating a complete loading of Ni to all bipyridine sites of MOF-253, leading to a formula of Al(OH)(dcbpy)(NiBr2) for MOF-NiBr2. TEM imaging and PXRD studies showed that MOF-NiBr2 maintained the needle-like morphology and crystallinity of MOF-253 (Figure 2b,d). X-ray photoelectron spectroscopy (XPS) spectrum in the Ni 2p region showed two main peaks, corresponding to spin-orbit coupled J=3/2 and 1/2 states, and two satellite peaks that are related to the shakeup of the two main peaks.19 The +2 oxidation state of Ni centers in MOF-NiBr2 was evident by the Ni 2p3/2 peak at ca. 855.0 eV (Figure S5b).20 N2 adsorption measurements gave a BET surface area of 499 m2/g for MOF-NiBr2 (Figure 2c), a value that is significantly smaller than that of pristine MOF-253 (1466 m2/g). The reduced porosity of MOF-NiBr2 can be attributed to the increased molecular weight after metalation and partial filling of the micropores by NiBr2.
MOF-NiBr2 was activated with excess NaBHEt3 to afford the active catalyst MOF-NiH (Figure 2a). The treatment of MOF-NiBr2 with NaBHEt3 led to a drastic color change from yellow to dark green and vigorous evolution of H2 bubbles. TEM and PXRD studies of MOF-NiH indicated the maintenance of needle-like morphology and crystallinity after NaBHEt3 treatment (Figure 2b, Figure 3a). N2 adsorption experiments gave a BET surface area of 364 m2/g for MOF-NiH, indicating the maintenance of high porosity (Figure S11). High-resolution TEM (HRTEM) image (Figure 3b) indicated that no Ni nanoparticles were formed during NaBHEt3 treatment. The generation of H2 during the treatment of MOF-NiBr2 with NaBHEt3 suggested bimetallic reduction elimination of H2 from the putative MOF-NiH2 to form MOF-NiH with (bpy)Ni(μ2-H)2Ni(bpy) active sites. Extended X-ray adsorption fine structure (EXAFS) spectroscopy confirmed the formation of (bpy)Ni(μ2-H)2Ni(bpy) coordination environments. Fitting of EXAFS results gave Ni−H and Ni−N distances of 1.69 and 1.99 Å, respectively. A strong scattering contribution from the neighboring Ni center was found in the secondary coordination sphere, with a Ni−Ni distance of 2.31 Å (Figure 3d).

(a) TEM image of MOF-NiH. (b) HRTEM image of MOF-NiH. (c) Ni 2p XPS spectrum of MOF-NiH. (d) EXAFS fitting of Ni K-edge adsorption of MOF-NiH. The inset shows the structure of (bpy)Ni(μ2-H)2Ni(bpy). (e) EPR spectrum of MOF-NiH (toluene, 15 K). (f) Proposed electronic structure of (bpy⋅−)NiII(μ2-H)2NiII(bpy⋅−) active sites in MOF-NiH.
Treatment of dark green MOF-NiH with excess formic acid changed the color of the solid to light green that is characteristic of NiII species, with concomitant formation of 3 equivalents of H2 for each (bpy)Ni(μ2-H)2Ni(bpy) active site (Figure S12). This result is consistent with protonation of both hydrides to form 2 equivalents of H2 and single-electron transfer from both Ni centers to two protons to form another equivalent of H2. However, XPS studies of MOF-NiH revealed a Ni oxidation state of +2 with the Ni 2p3/2 peak at ca. 855.8 eV (Figure 3c), which is close to the 2p3/2 peak of MOF-NiBr2 at ca. 855.0 eV. Electron paramagnetic resonance (EPR) spectrum of MOF-NiH (Figure 3e) showed a signal centered at g=2.008, indicative of an S=1/2 species. These results suggest the formation of (bpy⋅−)NiII(μ2-H)2NiII(bpy⋅−) active sites due to the redox non-innocence of bpy ligands (Figure 3f). The g-value measured for MOF-NiH corresponded to a free electron (ge=2.002), which supports the ligand-centered radicals delocalized in the bipyridine ligands.21 The generation of 3 equivalents of H2 for each dinickel site upon treatment of MOF-NiH with formic acid is consistent with the assignment of (bpy⋅−)NiII(μ2-H)2NiII(bpy⋅−) electronic structure. Although homogeneous [(α-diimine)Ni(μ2-H)]2 complexes with sterically hindered, redox non-innocent α-diimine ligands have been previously reported,21b, 22 a similar homogeneous complex with a bpy ligand is not known. Thus, site isolation in the MOF stabilizes the solution-inaccessible (bpy⋅−)NiII(μ2-H)2NiII(bpy⋅−) active sites.
MOF-NiH was tested for its catalytic activity in selective hydrogenation reactions. Considering the importance of alkyne semihydrogenation to produce (Z)-alkenes, diphenylacetylene was chosen as a model substrate to optimize the reaction conditions (denoted standard condition) for semihydrogenation of alkynes (Table 1). We chose n-hexane as the solvent based on its chemical inertness and lack of coordination ability.23 With 0.5 mol % MOF-NiH loading and 10 bar H2, 16 % of diphenylacetylene was converted after 72 h reaction in n-hexane at room temperature. Increasing the temperature to 50 °C led to the complete conversion of diphenylacetylene to afford (Z)-stilbene in 60 % yield along with the over-hydrogenated product 1,2-diphenylethane in 31 % yield. Decreasing the H2 pressure to 5 bar significantly increased the semihydrogenation selectivity, resulting in an 86 % conversion and a 73 % yield of (Z)-stilbene. The Z/E ratio of stilbene was determined as 24 by gas chromatography-mass spectrometry (GC-MS). Increasing the catalyst loading to 1 mol % led to a 100 % conversion of diphenylacetylene and an 82 % yield of (Z)-stilbene by GC-MS. Purification by column chromatography gave (Z)-stilbene as a colorless liquid in 80 % isolated yield.
|
|||||||||
Entry |
Catalyst |
Loading (mol %) |
H2 pressure (bar) |
Temperature (°C) |
Conversion (%) |
2 a (%)[b] |
3 a (%)[b] |
4 a (%)[b] |
Z/E |
|---|---|---|---|---|---|---|---|---|---|
1 |
MOF-NiH |
0.5 |
10 |
r. t. |
16 |
14 |
<1 |
2 |
– |
2 |
MOF-NiH |
0.5 |
10 |
50 |
100 |
60 |
9 |
31 |
6.7 |
3 |
MOF-NiH |
0.5 |
5 |
50 |
86 |
73 |
3 |
10 |
24 |
4 |
MOF-NiH |
1 |
5 |
50 |
100 |
82 (80[c]) |
3 |
15 |
26 |
5 |
MOF-253 |
2 |
5 |
50 |
0 |
0 |
0 |
0 |
– |
6 |
none |
– |
5 |
50 |
0[d] |
0 |
0 |
0 |
– |
7 |
NiBr2+2NaBHEt3[e] |
2 |
5 |
50 |
9[d] |
6[d] |
<1 |
3[d] |
– |
8 |
MOF-NiH1[e] |
2 |
5 |
50 |
<1[d] |
– |
– |
– |
– |
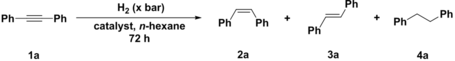
- [a] Reaction conditions: 1 a (0.25 mmol), H2, and catalyst (loadings based on dinickel sites) in 3 mL n-hexane for 72 h. [b] Conversions, yields, and Z/E ratios determined by GC-MS. [c] Isolated yield. [d] Determined by 1H NMR spectroscopy using CH2Br2 as an internal standard. [e] Loadings calculated based on mononuclear nickel sites.
The substrate scope of alkyne semihydrogenation was investigated using the optimized conditions. A variety of diaryl acetylenes were successfully hydrogenated to (Z)-stilbenes with MOF-NiH as the catalyst (Table 2). For substrates with alkyl substituents (−Me, −Et, and −tBu,), the desired (Z)-stilbenes were obtained in 74 %, 86 %, and 70 % at 50 °C. For the substrate with a strong electron-donating substituent (−OMe), the (Z)-stilbene was obtained in an 87 % yield at 35 °C. A lower temperature was needed to avoid overhydrogenation. For substrates with electron-withdrawing substituents (−Br, −Cl, −F, −COOMe, and −CF3), higher reaction temperatures were needed to produce the targeted (Z)-stilbenes in 89 %, 89 %, 62 %, 84 %, and 73 % yields, respectively. Cyclohexane was used as the solvent in these reactions due to its slightly higher boiling point than n-hexane.

- [a] Reaction condition: Substrate (0.25 mmol), H2 (5 bar), MOF-NiH (1 mol %) and solvent (3 mL) for 72 h. [b] Isolated yields. [c] Determined by GC-MS. [d] Solvent: n-hexane. [e] Solvent: cyclohexane. [f] Determined by 1H NMR spectroscopy using CH2Br2 as an internal standard.
Several control experiments were conducted to gain insights into the reaction mechanism of alkyne semihydrogenation. No conversion of the starting material was observed with the unmetalated MOF-253 as catalyst (Table 1, entry 5) or without catalyst (Table 1, entry 6). These results indicate that Ni sites in the metalated MOF are responsible for alkyne semihydrogenation. Only 9 % conversion was observed when in situ-generated Ni nanoparticles were used as the catalyst (Table 1, entry 7), suggesting the importance of the MOF support in catalysis. A mononuclear nickel hydride MOF, MOF-NiH1, was synthesized by metalation of MOF-253 with 0.12 equivalent of NiBr2(dme) and activation with NaBHEt3. The high crystallinity and porosity of MOF-NiH1 were confirmed by PXRD (Figure S15) and N2 adsorption (Figure S16) experiments, respectively. Under the standard conditions, dinuclear catalyst MOF-NiH gave 100 % conversion of the starting material, while mononuclear catalyst MOF-NiH1 with 2-fold loading of active sites gave less than 1 % conversion (Table 1, entry 8). This result shows the importance of the dinuclear Ni sites in the hydrogenation reaction.
Based on these experimental results and literature precedents on dicobalt-catalyzed alkyne semi-reduction,24 we proposed a plausible reaction mechanism for diphenylacetylene semihydrogenation in Figure 4a. First, diphenylacetylene coordinates to both Ni centers via a μ2,η2-binding mode. Migratory insertion into a Ni−H bond forms a Ni-bound 1,2-dipheylvinyl intermediate with syn stereochemistry. The Ni-vinyl intermediate reacts with H2 to afford (Z)-stilbene and regenerate the (bpy)Ni(μ2-H)2Ni(bpy) catalyst. The syn stereochemistry of the Ni-vinyl intermediate determines the (Z)-selectivity of the stilbene product.

(a) Proposed mechanism for MOF-NiH-catalyzed semihydrogenation of diphenylacetylene. (b) DFT-calculated energy profiles of MOF-NiH-catalyzed semihydrogenation of diphenylacetylene. (c) DFT-optimized structures of (bpy)Ni(μ2-H)2Ni(bpy) catalyst and intermediates.
We conducted density functional theory (DFT) calculations to support the proposed mechanism. The energy profile and optimized structures from DFT calculations are presented in Figure 4b and Figure 4c, respectively. Based on DFT calculations, the diphenylacetylene binding step and hydrogen insertion step are both energetically feasible, with ΔG values of −5.3 kcal/mol and 2.7 kcal/mol, respectively. Additionally, the ΔG in the third step, which involves the substitution of the formed diphenylethylene by H2, is −24.9 kcal/mol.
The recyclability of the MOF-NiH catalyst was also demonstrated. Five consecutive rounds of diphenylacetylene semihydrogenation were performed with an only slight decrease in (Z)-stilbene yields (Figure 5 and Table S2). Consistent with this, <0.01 % of Ni leaching was detected in the supernatant of the hydrogenation reaction. The PXRD pattern (Figure 2b) of the recovered MOF-NiH exhibited no discernible changes relative to the pristine MOF-NiH, suggesting that the MOF-NiH remained stable under catalytic conditions. The stability of MOF-NiH was further supported by our analysis of the recovered MOF-NiH using various techniques, including Fourier-transformed infrared spectroscopy (FTIR) (Figure S19), XPS (Figure S20), and TEM (Figure S21).

(Z)-stilbene yields (%) at different runs of MOF-NiH-catalyzed semihydrogenation of diphenylacetylene.
MOF-NiH also catalyzed selective hydrogenation of C=C bonds in α,β-unsaturated aldehydes and ketones. With 0.5 mol % MOF-NiH loading and 10 bar H2 in cyclohexane, trans-cinnamaldehyde was transformed at 90 °C to hydrocinnamaldehyde in 78 % yield over 48 h (Table 3, entry 1). Only the C=C bond was hydrogenated in the reaction, while the carbonyl group remained unchanged. Several α,β-unsaturated ketones were selectively hydrogenated in their C=C bonds in high yields (Table 3, entries 2–3). The electronic effects of the substituents followed a similar trend to alkyne semihydrogenation. Longer reaction times and higher temperature are needed for α,β-unsaturated ketones with the −Cl substituent (Table 3, entries 4 and 6). While 6 b was obtained in 96 % yield at 90 °C over 28 h, 6 d was obtained in 82 % yield over 48 h. A high turnover number of 192 was obtained for the selective hydrogenation of benzylideneacetone to 6 b. Similarly, while hydrogenation of trans-chalcone at 90 °C for 48 h gave 6 e in 79 % yield (Table 3, entry 5), hydrogenation of p-chlorochalcone was carried out at 120 °C to afford 6 f in 61 % yield (Table 3, entry 6).

- [a] Reaction condition: substrate (0.50 mmol), H2 (10 bar), MOF-NiH (0.5 mol %) and cyclohexane (3 mL) for 28 to 48 h. [b] Isolated yields. [c] Determined by 1H NMR spectroscopy using CH2Br2 as an internal standard.
Conclusion
In this work, we have synthesized a novel MOF-based dinuclear NiII2(μ2-H)2 catalyst for semihydrogenation of alkynes to (Z)-alkenes and selective hydrogenation of C=C bonds in α,β-unsaturated aldehydes and ketones. Spectroscopic studies identified solution-inaccessible (bpy⋅−)NiII(μ2-H)2NiII(bpy⋅−) as the catalytically active site. Selective hydrogenation products were obtained in high isolated yields with turnover numbers of up to 192. The present work highlights the potential of generating unusual and solution-inaccessible bimetallic catalysts in MOFs for sustainable catalysis and fine chemical synthesis.
Supporting Information
The authors have cited additional references within the Supporting Information.25
Acknowledgments
We thank Dr. Dong Won Kang, Dr. Haifeng Zheng, Prof. John Anderson, Dr. Lei Wang, Joseph Schneider, Jiuyun Shi, Chen-Yu Lien, and Ziwan Xu for experimental help. We thank Dr. Alexander Filatov for help with XPS and PXRD measurements. We acknowledge the National Science Foundation (CHE-2102554) and the University of Chicago for funding support and the MRSEC Shared User Facilities at the University of Chicago (DMR-1420709) for instrument access. XAS analysis was carried out at Beamline 10-BM, supported by the Materials Research Collaborative Access Team (MRCAT). Use of the Advanced Photon Source, an Office of Science User Facility operated for the U.S. DOE Office of Science by ANL, was supported by the U.S. DOE under Contract DE-AC02-06CH11357.
Conflict of interest
The authors declare no conflict of interest.
Open Research
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.

