

Feinjustierung von Substrat–Katalysator Halogen–Halogen Interaktionen zur Steigerung der Enantioselektivität in der Halogenbrückenbindungs-Katalyse
Abstract
Ein neuer Ansatz für eine hoch enantioselektive Halogenbrückenbindungs-Katalyse wurde entwickelt. Um die intrinsischen Probleme der Halogenbrückenbindungen (XB) und die daraus resultierenden ungelösten Limitierungen in der asymmetrischen Katalyse zu umgehen, wurden fein justierte Halogen–Halogen Interaktionen zwischen dem Substrat und dem XB-Donor designt, um das Substrat im Katalysatorhohlraum vorzuorganisieren und die Kontrolle über die Enantioselektivität zu verbessern. Bei der vorliegenden Strategie wurden sowohl die Elektronenwolke (Lewis basische Stelle) als auch die sigma (σ)-Loch Stelle des Halogen-substituenten der Substrate genutzt, um einen engen chiralen Katalysator–Substrat–Gegenanion Komplex zu bilden. Dies ermöglicht eine kontrollierte Einführung eines hohen Chiralitätstransfers. Bemerkenswerte Enantioselektivitäten von bis zu 95 : 5 e.r. (90 % ee) wurden erreicht in einer Modell-Dearomatisierungsreaktion von halogensubstituierten (Iso)chinolinen mit mehrzähnigen anionen-bindenden Tetrakis-Iodotriazol Katalysatoren.
Einleitung
In den letzten Jahren haben sich Halogenbrückenbindungen1 als leistungsstarkes Werkzeug in der supramolekularen2 und medizinischen Chemie3 erwiesen und neue Möglichkeiten in der selektiven Erkennung4 und Organokatalyse5, 6 geschaffen. Wichtige Beiträge zur enantioselektiven Erkennung von neutralen und anionischen Substraten wurden unter anderem mit verschiedenen chiralen Rotaxanen,7 Halotriazol- und Halotriazolium-basierten Halogenbrückenbindungs (XB)-Donoren8 geleistet. Die meisten der bisherigen XB-Prozesse basieren jedoch auf achiralen Donoren,1 und enantioselektive Anwendungen,7, 8 insbesondere in der Katalyse,9, 10-13, 14, 15 stecken noch in den Kinderschuhen. In dieser Hinsicht gibt es einige Herausforderungen, die überwunden werden müssen, um XB in enantioselektiven Prozessen wirkungsvoll einzusetzen. So führt die hohe Direktionalität von etwa 180° zwischen dem sigma (σ)-Loch des Halogens und dem gebundenem Substrat (Lewis Base = LB) sowie die Größe des Halogenatoms, in der Regel Iod, zu einem längeren Abstand zwischen dem chiralen Grundkörper des Katalysators und dem Substrat, wodurch ein hoher Chiralitätstransfer schwieriger zu erreichen ist. Aus diesem Grund gibt es bisher nur wenige Beispiele für chirale XB-Donoren, die in der asymmetrischen Katalyse eingesetzt werden.
Die ersten enantioselektiven Ansätze wurden von den Gruppen von Tan11 und Arai12 beschrieben, die XB in der dualen Katalyse als sekundäre Interaktion zwischen mehreren nicht-kovalenten und/oder Lewis-Säure/Base Aktivierungseinheiten verwendeten. Interessanter, aber auch anspruchsvoller ist die Verwendung von XB und generell der σ-Loch Aktivierung als primäre Wechselwirkung, welche in der asymmetrischen Katalyse noch unterrepräsentiert ist. 2020 veröffentlichte die Gruppe von Huber das erste Beispiel für eine asymmetrische Induktion, die ausschließlich auf einer XB-Aktivierung mit einem chiralen zweizähnigen Bis-Iodoimidazolium XB-Donor, der große Seitenarme trägt, um eine chirale Umgebung in der Nähe des Substrates zu schaffen (Abbildung 1a, links).14 In einer Mukaiyama-Aldol Reaktion mit Glyoxal-Derivaten konnte eine Enantioselektivität von bis zu 33 % ee erreicht werden. Kurz darauf veröffentlichte unsere Gruppe ein chirales Tetrakis-Iodotriazol System15 für die enantioselektive Anionenbindungskatalyse,18 das eine vergleichbar geringe Enantioinduktion in einer Reissert-Typ Reaktion erreichte (Abbildung 1a, rechts).15, 19 Die in dieser Studie durchgeführten quantenchemischen Berechnungen zeigten eine zweizähnige Bindung von nur einem Katalysatorarm an das Chlorid im Katalysator Substrat Ionenpaar-Komplex.

a) Stand der Technik bei der Enantioinduktion in der XB-Katalyse. b) Diese Arbeit: Feinjustierung der Substrat–Katalysator Halogen-Interaktionen mit Triazol-XB-Donoren zur Verbesserung der Chiralitätsinduktion in der XB-Anionenbindungskatalyse, unterstützt durch quantenchemische Berechnungen.
Um einige der gegenwärtigen Limitierungen in der asymmetrischen σ-Loch Katalyse zu lösen, zielten wir auf eine bessere Fixierung des Substrates und damit auf einen effizienteren Chiralitätstransfers ab, um synthetisch nutzbare Enantioselektivitäten zu erreichen. Aus diesem Grund haben wir uns zusätzliche Substrat–Katalysator Halogeninteraktionen als Strategie zur Steigerung der Enantioselektivität in der XB-Katalyse überlegt. Als Beweis für dieses Prinzip untersuchten wir diesen Ansatz in einer ähnlichen, durch Anionenbindung katalysierten prototypischen Reissert-Typ Reaktion (Abbildung 1b). Im Speziellen haben wir N-Arenium Substrate vorgeschlagen, die zusätzliche Halogene (X) tragen, die gleichzeitig als Lewis-Basen durch ihre Elektronenwolke (LB-Stelle, in blau) und als σ-Loch Donor (XB-Stelle, in dunkelviolett) wirken können, um fein justierte multiple Halogen–Halogen Kontakte mit dem XB-Katalysator zu ermöglichen. DFT-Berechnungen des plausiblen Übergangszustands (TS) eines Modellsystems haben gezeigt, dass das Zusammenspiel von Halogeninteraktionen ein Schlüsselelement für ein hohes Maß an Enantiokontrolle ist, und legen die Beteiligung von vier σ-Loch Interaktionen mit dem Chlorid-Gegenion des Substrates und zwei Halogen–Halogen Kontakten zwischen dem Substrat und dem Katalysator nahe. Basierend auf dieser neuen Strategie in der XB-Katalyse beschreiben wir hier die asymmetrische Dearomatisierung von halogensubstituierten (Iso)chinolinen mit anionenbindenden Tetrakis-Iodotriazol Katalysatoren, wobei hohe Enantioselektivitäten von bis zu 95 : 5 e.r. (≤90 % ee) erreicht werden.
Ergebnisse und Disskusion
Optimierung der Reaktion: Halogen-Substitutionseffekt
Um unsere Hypothese zur Verbesserung der Stereokontrolle durch eine zusätzliche Halogeninteraktion zwischen dem Katalysator und dem Substrat, das eine Lewis basische Stelle am Gerüst trägt, zu untersuchen, wurden zunächst mehrere Halochinoline getestet (Schema 1). Nach der üblichen Präaktivierung mit TrocCl zu den N-Acylchinoliniumchlorid-Salzen wurde die Reaktion mit dem Silylketenacetal 3 a in CH3Cl bei −60 °C in Gegenwart von 5 Mol % Tetraiodotriazol Katalysator 1 a durchgeführt (Methode A). Durch die Rotation eines Chloratoms um das Chinolingerüst konnte ein erheblicher Einfluss auf das Ergebnis der Enantioselektivität der Reaktion beobachtet werden. Das beste Ergebnis wurde mit 4-Cl-Chinolin erzielt, welches das Produkt 4 aa in einer exzellenten Ausbeute von 99 % und einem guten Enantiomerenverhältnis von 71 : 29 e.r. zugunsten des (S)-Enantiomers lieferte. Andere Substitutionsmuster, wie z. B. 3-Cl (2 ab), 5-Cl (2ad), 6-Cl (2 ae) und 7-Cl-Chinolin (2 af), führten zu verringerten Enantioinduktionen (≤60 : 40 e.r.), die sogar geringer waren als beim unsubstituierten Ausgangs Chinolin (4 a). Darüber hinaus wurde bei 2-Cl (2 ac) und 8-Cl-Chinolin (2 ag) keine Reaktion beobachtet. In diesen beiden Fällen könnte der Substituent den nukleophilen Angriff behindern, indem er die C-2-Position direkt blockiert bzw. die freie Drehung der N-Troc-Schutzgruppe einschränkt.

Screening der Wirkung des Einbaus eines Halogensubstituenten in das Substrat 2. Alle Reaktionen wurden im 0.1 mmol-Maßstab in dem entsprechenden Lösungsmittel (0.05 M; Methoden A–C) unter Argonatmosphere durchgeführt. Isolierte Ausbeuten gegeben. E.r. bestimmt durch chirale SFC oder HPLC-Analyse.
Weiter wurde die Auswirkung von schwereren Halogen-substituenten in der 4-Position untersucht. Wir waren erfreut, dass die Enantioselektivität beim Übergang von einer 4-Cl- zu einer 4-I-Substitution zunahm, was auf eine stärkere Halogenbrückenbindung und/oder eine bessere Interaktion mit dem weicheren, stärker polarisierten Iodatom hinweisen könnte. Die dearomatisierten Produkte 4 ba und 4 ca wurden in 75 : 25 e.r. erhalten, allerdings nur in moderater Ausbeute (34 bzw. 37 %). Das war das Ergebnis einer kompetitiven Halogenaustausch Nebenreaktion mit dem vorliegenden Cl-Gegenanion des N-Troc-Chinolinium Substrats. Um diesen Prozess zu vermeiden oder minimieren, wurde das Lösungsmittel zu Et2O gewechselt (Methode B). Erfreulicherweise wurden die gewünschten Produkte dann selektiv (>90 %) und mit leicht verbesserter Enantioselektivität erhalten, wobei für 4 ca mit der 4-Iod-Substitution 78 : 22 e.r. erreicht wurde.
Um die Enantioselektivität weiter zu verbessern und gleichzeitig den I-Cl-Austausch zu unterdrücken, wurde ein Lösungsmittel-Screening mit 4-Iodochinolin (2 ca) durchgeführt (siehe Hintergrundinformationen für das vollständige Screening).
Es stellte sich heraus, dass das beste Lösungsmittelsystem eine 2 : 1 Mischung aus Et2O/C6F6 bei −40 °C (Methode C) war, die das Produkt 4 ca in 56 % Ausbeute und einem deutlich verbesserten Verhältnis von 83 : 17 e.r. lieferte, während ein geringer oder nicht feststellbarer Halogenaustausch zum 4-Chlor-Nebenprodukt stattfand (4-I/4-Cl-Selektivität >95 %).
Berechnungsstudie: Substrat-Katalysator-X-Interaktion
Aufgrund unserer ersten Beobachtungen und um weitere Einblicke in die Schlüsselinteraktionen zu gewinnen, die zur Verbesserung der Enantioselektivität durch die Einführung von Halogenatomen im Substrat führen, wurden Berechnungen auf Dichtefunktionaltheorie (DFT) Ebene durchgeführt. Im Speziellen wurde das M06-2X20-Funktional mit den Basissätzen 6-31G(d,p)21 und LANL2DZ22 für die Iodatome zur Optimierung mit zusätzlichen Lösungsmittel- und Energiekorrekturen verwendet (siehe Hintergrundinformationen für weitere Einzelheiten).
Der Katalysator 1 a und 4-Iodochinolin (2 ca) wurden zusammen mit einem Modell Silylketenacetal (((1-Methoxy-vinyl)oxy)trimethylsilan) als Nukleophil verwendet, um die Halogeninteraktionen zwischen dem XB-Katalysator und dem Substrat entlang des Reaktionsweges zur Bildung von (S)- und (R)-Produkten zu untersuchen (siehe Hintergrundinformationen für ein vollständiges Bild). Unter zehn verschiedenen Reaktionswegen ist einer der wahrscheinlichsten Wege zur Bildung des beobachteten (S)-Hauptprodukts einer der mehrfache XB-induzierte Stabilisierungen im Übergangszustand ts1S aufweist (Abbildung 2). Im Gegensatz zu dem zuvor beobachteten zweizähnigen Modus (siehe Abbilding 1a, rechts)15 weist der Katalysator 1 a eine dreizähnige Bindung vom Typ XB an das Chloridanion in ts1S auf. Zudem zeigt das acylierte Iodochinolinium-Substrat, das auch als schwacher Halogenbrückendonor fungieren kann, eine zusätzliche XB zum Chloridanion (mit einer NBO-Stabilisierungsenergie von 13.7 kcal mol−1). Dieser XB-Kontakt durch das σ-Loch des Substrates wird vermutlich durch I–I-Interaktionen mit zwei Iodotriazolen von 1 a (Typ I,24 mit Winkeln von θ1=134° und θ2=113° mit ML-I, und θ1=131° und θ2=114° mit RA-I) verstärkt, die ähnliche NBO-Stabilisierungsenergien von 0.8 bzw 0.7 kcal mol−1 aufweisen. Die Stabilisierung der XB zwischen dem äußeren linken Iodotriazol (LA-I) und dem Chlorid ist deutlich schwächer als für die anderen Iodo-Einheiten des Katalysators (ML-I und RA-I) und des Substrates (7.9 vs. ≈12–14 kcal mol−1). Diese Beobachtung könnte als treibende Kraft für den dreizähnigen Bindungstypen des Katalysators an das Chlorid in ts1S gedeutet werden. Somit spielen die zusätzlichen XB zwischen dem Substrat und dem Katalysator-Anion-Komplex eine entscheidende Rolle, um eine bessere Substratfixierung und somit einen effizienteren Chirali-tätstransfer zu erreichen, der den (S)-Produktbildungsweg begünstigt und zu den beobachteten erhöhten Enantioselektivitäten führt.

a) Molekulares elektrostatisches Potenzial (MEP) des Schlüsselübergangszustandes ts1S; b) dargestellte kooperative XB- und Halogen–Halogen-Interaktionen zwischen Katalysator 1 a, Chloridanion und Substrat in ts1S; MEP und Stabilisierungsenergien in kcal mol−1.
Katalysator Screening
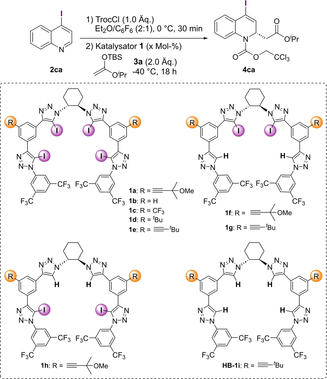
Motiviert durch die erzielten Ergebnisse und Erkenntnisse über das Halogen-Interaktionsnetzwerk wurden als nächstes weitere Triazol-basierte XB-Katalysatoren 1 in der Modellreaktion von 2 ca in Et2O/C6F6 bei −40 °C untersucht (Tabelle 1). Zunächst wurde eine Reihe von Tetraiodo XB-Donoren 1 a–e eingesetzt, die unterschiedlichen Reste an der Seitenkette tragen. Während die fehlende Seitenkettensubstitution zu einer geringeren Enantio-induktion führte (1 b: 71 : 29 e.r. vs. 1 a: 83 : 17 e.r., Einträge 1 und 2), bieten andere Gruppen wie CF3 oder tBu eine ähnlich gute Enantioselektivität (1 c: R=CF3, 80 : 20 e.r.; 1 d: R=tBu, 84 : 16 e.r., Einträge 3 bzw. 4). Das beste Ergebnis wurde mit dem Tetraiodo XB-Donor 1 e erzielt, der einen tert-Butyl-Acetylen Rest trägt, und 85 : 15 e.r. erreichte (Eintrag 5).
|
||||
Eintrag |
Katalysator (Mol %) |
Ausbeute [%][b] |
Selektivität [%] (4-I vs. 4-Cl)[c] |
e.r.[d] |
|---|---|---|---|---|
1 |
1 a (5) |
56 |
96 |
83 : 17 |
2 |
1 b (5) |
68 |
95 |
71 : 29 |
3 |
1 c (5) |
66 |
96 |
80 : 20 |
4 |
1 d (5) |
67 |
96 |
84 : 16 |
5 |
1 e (5) |
60 |
96 |
85 : 15 |
6 |
1 f (5) |
54 |
92 |
85 : 15 |
7 |
1 g (5) |
54 |
90 |
81 : 19 |
8 |
1 h (5) |
60 |
92 |
21 : 79 |
9 |
HB-1 i (5) |
77 |
90 |
21 : 79 |
10 |
1 e (2.5) |
80 |
95 |
86.5 : 13.5 |
11 |
1 e (10) |
67 |
95 |
84 : 16 |
- [a] Alle Reaktionen wurden im 0.1 mmol-Maßstab in trockenem Et2O/C6F6 (2 : 1, 0.05 M) und unter Argonatmosphäre durchgeführt. [b] Isolierte Ausbeuten. [c] Bestimmt durch NMR des Rohproduktes. [d] Bestimmt durch chirale SFC-Analyse.
Als nächstes wurden Bisiodotriazole an den “inneren” (1 f–g) oder “äußeren” Positionen (1 h) getestet. Ähnliche Enantioselektivitäten wurden mit 1 f und 1 g beobachtet (85 : 15 and 81 : 19 e.r., Einträge 6 und 7), wobei in diesem Fall die Dimethylmethoxy-Substitution an der Acetylen Seitenkette überlegen war. Interessanterweise wurde mit dem “äußeren” Bisiodotriazol Katalysator 1 h das entgegengesetzte (R)-Enantiomer bevorzugt, was den Ergebnissen ähnelt, die mit den Proto-Tetrakis-Triazol Systemen erzielt wurden (21 : 79 e.r., Einträge 8 und 9).19 Dies deutet darauf hin, dass das Vorhandensein von zwei großen Iodatomen an den “inneren” Positionen des Katalysators die Bildung des gleichen helikalen Katalysator-Anion-Komplexes behindert. Um eine starke Konkurrenz mit H-Bindungen bei den gemischten Bisiodo-Systemen zu vermeiden wurde 1 e als XB-Donor der Wahl identifiziert und die Katalysatormenge untersucht. Während die Verwendung von nur 2.5 Mol % 1 e zu einer erhöhten Ausbeute von 80 % und 86.5 : 13.5 e.r. führte (Eintrag 10), brachte die Verwendung von 10 Mol % Katalysatormenge keine Verbesserung (Eintrag 11). Daher wurden weitere Reaktionen mit 2.5 Mol % 1 e im optimierten Lösungsmittelsystem durchgeführt.
Bandbreite der Reaktion mit 4-Iodochinolinen
Wir setzten die Studie fort, indem wir die Bandbreite und die Grenzen der Methode untersuchten (Schema 2). Zunächst wurden unterschiedliche Silylketenacetal-Nukleophile untersucht. Unter Beibehaltung der TBS-Gruppe, lieferte der sterisch anspruchsvollere tBu-Rest in 3 b eine deutlich geringere Ausbeute und Enantioselektivität im Vergleich zum Ausgangsnukleophil 3 a mit einer iPr-Substitution (4 cb: 29 %, 72 : 18 e.r., vs. 4 ca: 80 %, 86.5 : 13.5 e.r.). Im Gegensatz dazu brachten die linearen Alkylgruppen bessere Ergebnisse. Mit Ausnahme des Ethylderivats 3 d (4 cd: 85.5 : 14.5 e.r.) führte die Methyl-, n-Propyl- und n-Butyl-Substitution zu einer Verbesserung der Enantioselektivität und erreichte in allen Fällen ein Verhältnis von 88 : 12 e.r. Das kommerziell erhältliche OMe-Nukleophil 3 c war jedoch reaktiver und bildete das Produkt 4 cc in einer hohen Ausbeute von 93 %. Abschließend ergaben das dimethylsubstituierte Trimethyl-silylketenacetal 3 g und das TBS-Dienolat 3 h die entsprechenden Produkte 4 cg und 4 ch in mäßiger bis hoher Ausbeute (70–93 %), jedoch mit deutlich niedrigeren Enantiomereninduktionen (81.5 : 18.5 e.r. bzw. 69 : 31 e.r.). Als nächstes wurde der Substratumfang für eine Reihe substituierter 4-Iodochinoline mit dem Nukleophil 3 c untersucht. Die Anwesenheit eines zweiten Halogens in C6- und C7-Position war kompatibel und führte zu den Produkten (4 ci–4 cn) in mäßigen bis guten Ausbeuten und ähnlichen Enantioselektivitäten wie für das Ausgangssubstrat 2 ca (81 : 19–88 : 12 e.r.). Umgekehrt zeigte das C5-Fluoroderivat keine Reaktivität, was auf ineffiziente zusätzliche XB-Interaktionen des benachbarten C4-Iodatoms im Übergangszustand hindeuten könnte. Darüber hinaus wurden sowohl elektronenziehende Substituenten wie Trifluoromethyl (4 cp) als auch elektronendonierende Gruppen wie Methyl (4 cq und 4 cs) oder Methoxy (4 cr und 4 ct) gut toleriert und lieferten ein vergleichbares Maß an Enantioinduktion. Interessanterweise wurden die Pinacolboran-Produkte 4 cu und 4 cv, die orthogonale Kreuzkupplungsreaktionen ermöglichen, in guten Ausbeuten und hohen Enantioselektivitäten von bis zu 88 : 12 e.r. erhalten. Schließlich ist zu bemerken, dass die Reaktion mit Pyridinen wie 4-Iod-2-methyl-pyridin ebenfalls reibungslos verlief und das entsprechende Produkt 4 cw in 76 % Ausbeute und 81 : 19 e.r. lieferte.

Umfang der Reaktion mit Chinolinen 2. Alle Reaktionen wurden im 0.1 mmol-Maßstab durchgeführt. Isolierte Ausbeuten angegeben. Der e.r. wurde durch chirale SFC oder HPLC-Analyse bestimmt. Die Selektivität der 4-I/4Cl-Produktbildung wurde durch 1H NMR des Rohproduktes bestimmt.
Die Robustheit der Methode wurde durch eine 10-fache Hochskalierung der Reaktion von 2 ca mit 3 c bestätigt (Abbildung 3a). Unter Standardbedingungen lieferte die Reaktion von 2 ca (255.5 mg, 1.0 mmol) mit 3 c das Produkt in ähnlich hoher Ausbeute (90 % vs. 93 % für 0.1 mmol) und demselben Enantiomerenverhältnis von 88 : 12 e.r.

a) Hochskalierung der Reaktion 2 ca mit 3 c, und b) Vergleich der Wirkung verschiedener C4-substituierter Chinoline.
Um besser differenzieren zu können, welche Rolle die 4-Iodogruppe des Substrates als σ-Loch-Donor oder Lewis-basische Stelle spielt, wurden schließlich unterschiedliche C4-substituierte Chinoline untersucht (Abbildung 3b). Die Reaktion wurde mit einem Chinolin durchgeführt, das eine Phenyltelluriumgruppe enthält, die in der Lage ist, Chalkogenbrückenbindungen (ChB) mit dem Katalysator oder dem anionen-bindenden Komplex einzugehen. In diesem Fall wurde das Produkt 4 cf in 75 % Ausbeute und einem ähnlichen Verhältnis von 84 : 16 e.r. gebildet. Umgekehrt lieferte eine Methoxygruppe, die als Lewis-Base fungieren kann, aber kein guter σ-Loch-Donor ist, oder eine 4-Methyl-Substitution die Produkte 4 ec und 4 dc mit deutlich geringeren Enantioselektivitäten (63 : 37 e.r.), vergleichbar mit der des unsubstituierten Chinolins. Diese Beobachtungen stehen im Einklang mit den Berechnungen (siehe ts1S, Abbildung 2) und deuten darauf hin, dass das katalytische System nicht nur mit Halogenbrückenbindungen arbeitet, sondern auch auf andere σ-Loch-Interaktionen wie Chalkogenbrückenbindungen übertragen werden kann.
Erweiterung auf Isochinoline
Als nächstes erweiterten wir unseren Substrat-Katalysator-Halogen-Interaktionsansatz für die Reaktion mit den verwandten Isochinolinen (Schema 3). Auch hier wurde die optimale Position des zusätzlichen Halogens im Substrat durch Rotation eines Bromatoms am Isochinolingerüst untersucht. Erfreulicherweise stellte sich diese Art von Substraten als noch geeigneter heraus, da diese die dearomatisierten Produkte im Allgemeinen in höheren Enantiomerenverhältnissen liefern als die Ausgangschinoline. Das beste Ergebnis lieferte das 5-Bromoderivat (6 aa: 60 %, 94.5 : 5.5 e.r.), gefolgt von den 4-Bromo- und 6-Bromo-isochinolinen (6 ae: 81 %, 92.5 : 7.5 e.r.; 6 ab: 7 %, 91 : 9 e.r.), während die 7- und 8-Substitutionen 6 ca und 6 ad nur geringe Enantiomereninduktionen lieferten. Auf Grundlage dieser Ergebnisse wurden auch 5-Chlor (5 b) und 5-Iod (5 c) untersucht. In diesem Fall konnte nur ein vernachlässigbarer Unterschied zwischen der Chlor-, Brom- und Iod-Substitution festgestellt werden (im Bereich von 93 : 7 bis 94.5 : 5.5 e.r.). Außerdem ist zu bemerken, dass der Halogenaustausch zu den 5-Chloroisochinolin Produkten im Vergleich zu den 4-Halochinolin Produkten äußerst gering ist. Darüber hinaus wurde eine verminderte chirale Induktion mit dem 5-Methoxyisochinolin (6 da: 69 : 31 e.r.) erzielt. Dies steht im Einklang mit unserer Hypothese, dass zusätzliche Halogeninteraktionen zwischen dem Substrat und dem Katalysator-Anion-Komplex erforderlich sind, um eine hohe Enantioselektivität zu erreichen.

Umfang der Reaktion mit Haloisochinolinen 5.
Im Hinblick auf ihre bessere Verfügbarkeit wurden letztlich einige 5-Bromoisochinoline, die einen zusätzlichen elektronenziehenden, elektronendonierenden oder neutralen Substituenten an der C6-Position enthalten, unter den Standardbedingungen umgesetzt. Die 5-Bromo-6-methyl und -6-Methoxy-Derivate lieferten die Produkte 6 ag und 6 ah mit etwas geringerer Enantioselektivität als das 5-Bromoisochinolin (85 : 15 e.r. bzw. 83 : 17 e.r.). Zu unserer Freude ergab die Einführung eines Fluors an der C6-Position ein ausgezeichnetes Enantiomerenverhältnis von 95 : 5 (6 af).
Zusammenfassung
Zusammenfassend lässt sich sagen, dass das seit langem bestehendem Problem des ineffizienten Chiralitätstransfers bei der Halogenbrückenbindungs-Katalyse zum Teil durch die Einführung fein justierter Halogen–Halogen-Interaktionen zwischen dem Substrat und dem XB-Donor gelöst werden konnte. Im Rahmen dieses Ansatzes wurde eine asymmetrische Dearomatisierungsreaktion von halogensubstituierten N-Heteroarenen wie (Iso)chinolinen unter Verwendung von Tetrakis-Iodotriazolen als mehrzähnige anionenbindende Katalysatoren untersucht. Berechnungen ergaben, dass das Substrat sowohl über seine Elektronenwolke (Lewis basische Stelle) als auch über die σ-Loch-Stelle zusätzliche Halogeninteraktionen mit dem Chlorid–Katalysator-Komplex eingeht. Darüber hinaus wurden auch verbesserte Halogenbrückenbindungs-Eigenschaften durch Iod–Iod-Interaktionen festgestellt. Abschließend unterstreicht der Vergleich mit Substraten, die eine MeO- (Lewis-Base) oder eine PhTe-Gruppe (σ-Loch-Donor) enthalten, die entscheidende Rolle einer σ-Loch-Brücke des Substrats für eine effiziente Enantioinduktion. Somit konnten hohe Enantioselektivitäten von bis zu 95 : 5 e.r. erreicht werden, die weit über den derzeitigen Stand der Technik in der XB-Katalyse hinausgehen.
Danksagung
Dem European Research Council (ERC-CG 724695) und der Deutschen Forschungsgemeinschaft (DFG) wird im Rahmen des IRTG2027 für die großzügige Unterstützung gedankt. D.G.P. dankt der Europäischen Union und dem polnischen Ministerium für Wissenschaft (Nr. 847413) sowie dem polnischen Hochschulwesen (5005/H2020-MSCA-COFUND/ 2019/2). Wir danken auch für die großzügige Bereitstellung von Computerzeit an den Hochleistungsrechenzentren: PALMA an der WWU Münster und ICM an der UW Warschau. Open Access Veröffentlichung ermöglicht und organisiert durch Projekt DEAL.
Interessenkonflikt
Die Autoren erklären, dass keine Interessenkonflikte vorliegen.
Open Research
Data Availability Statement
Die Daten, die die Ergebnisse dieser Studie unterstützen, sind in den Hintergrundinformationen zu diesem Artikel verfügbar.

