

Wacker Oxidation von Methylencyclobutanen: Umfang und Selektivität in einer ungewöhnlichen Umgebung
Abstract
Methylencyclobutane werden der Wacker Oxidation über eine semi-pinakolartige Umlagerung unterzogen. Der Schlüssel zu einem erfolgreichen Prozess ist die Verwendung von tert-Butylnitrit als Oxidationsmittel, das nicht nur eine effiziente Katalyse ermöglicht, sondern auch eine hohe Markovnikov-Selektivität unter milden Bedingungen gewährleistet. Auf diese Weise können Cyclopentanone (26 Beispiele) in guter Ausbeute und ausgezeichneter Selektivität (bis zu 97 % Ausbeute und generell >99 : 1 Keton:Aldehyd-Verhältnis) zugänglich gemacht werden. Die stereochemische Analyse der Reaktionssequenz zeigt ein Wanderungsverhalten, das mit verwandten 1,2-Umlagerungen übereinstimmt. Des Weiteren können durch die Einführung eines Pyox-Liganden an Palladium prochirale Methylencyclobutane desymmetrisiert werden, was der ersten enantioselektiven Wacker-Oxidation entspricht.
Einleitung
Seit ihrer Entdeckung im Jahre 19561 stellt die Wacker Oxidation eine unverzichtbare Methode zur Umwandlung von Olefinen in Carbonyle dar.2, 3 Traditionell wird Palladiumchlorid als Katalysator verwendet, während Kupferchlorid die aerobe Reoxidation im wässrigen Reaktionsmedium vermittelt. Die Sequenz funktioniert besonders gut bei monosubstituierten Alkenen, die je nach Substratklasse und Reaktionsbedingungen entweder ketonselektiv4 oder aldehydselektiv5, 6 oxidiert werden können (Schema 1, oben).7 Interne Alkene können ebenfalls adressiert werden, auch wenn regioselektive Protokolle seltener sind.8 Im Gegensatz dazu ist die Wacker Oxidation von 1,1-disubstituierten Alkenen weitgehend unerforscht, mit Ausnahme des Palladium-katalysierten intra-molekularen Ringschlusses von anhängenden Hydroxygruppen. Dieser Prozess wird allgemein als Wacker-Zyklisierung bezeichnet (Schema 1, Mitte).9 Während einige wenige Berichte die klassische Wacker-Oxidation von 1,1-disubstituierten Alkenen zu Aldehyden beschreiben,10 sind Ketone aufgrund eines fehlenden β-Wasserstoffatoms im Allgemeinen nicht über diese Sequenz zugänglich. Wir haben jedoch vermutet, dass durch Einbeziehen einer formalen 1,2-Kohlenstoffverschiebung in die Wacker Oxidation ein umgelagertes Keton aus einem 1,1-disubstituierten Alkenvorläufer zugänglich wird (Schema 1, unten). In diesem Forschungsartikel fassen wir unsere Ergebnisse zusammen, die von der oben genannten Hypothese bis zur Entwicklung des ersten hocheffizienten Prozesses reichen.

Aktueller Umfang und Limitierungen der Wacker Oxidation. FG=funktionelle Gruppe.
Unsere detaillierte Hypothese für den Zugang zu umgelagerten Ketonen ist in Schema 2 dargestellt. Auf der Grundlage der zahlreichen mechanistischen Studien zur Hydroxypalladierung von monosubstituierten Alkenen,11 nahmen wir an, dass ein ähnlicher Prozess auch für 1,1-disubstituierte Alkene möglich sein sollte, der zu einem entsprechenden Zwischenprodukt vom Typ I-1 führt. Obwohl dieser Prozess reversibel und im Allgemeinen nicht produktiv ist, weist I-1 einige interessante Ähnlichkeiten mit den Substraten von Semipinakol-Umlagerungen auf, nämlich einem tertiären Alkohol mit PdII als latenter Abgangsgruppe in einer 1,2-Beziehung. Daher kann eine konzertierte 1,2-Verschiebung zum umgelagerten Keton vom Wacker-Typ erwartet werden (Schema 2, oben). Alternativ ist auch eine schrittweise 1,2-Verschiebung durch β-Kohlenstoff-Eliminierung und Wiedereinbau über das Alken denkbar (Schema 2, unten).

Mechanistische Hypothese für die Schlüsselumlagerung der Wacker Oxidation von 1,1-disubstituierten Alkenen.
Um eine solche Sequenz durchzuführen, wurden Methylencyclobutane (MCBs) als ideale Substrate ins Auge gefasst, da es eine Fülle von Palladium katalysierten 1,2-Verschiebungen von Alkenyl- und Alkinylcyclobutanolen zu den entsprechenden Cyclopentanonen gibt.12, 13, 14 Darüber hinaus ist bekannt, dass stöchiometrisches PdII-Nitrit15 die oxidative Ringerweiterung von MCB zu Cyclopentanon ermöglicht. Dies ist ein weiteres Indiz dafür, dass, sofern ein geeignetes Oxidationsmittel identifiziert werden kann, eine Wacker-Reaktion dieses Typs von 1,1-disubstituierten Alkenen möglich ist. Erste Hinweise auf einen solchen katalytischen Prozess wurden 1977 von Boontanonda und Grigg veröffentlicht, die eine oxidative Ringerweiterung für drei Substrate beschrieben.16
Ergebnisse und Diskussion
Wir begannen unsere Untersuchung zur Identifizierung geeigneter Wacker-Bedingungen für die MCB-Oxidation, indem wir 3,3-Diphenylmethylencyclobutan (1 a) als Modellsubstrat verwendeten (Tabelle 1 zeigt unsere wichtigsten Ergebnisse, für die vollständige Optimierung siehe Hintergrundinformationen). Unter klassischen Wacker-Tsuji-Bedingungen (Eintrag 1) wurde keine Umwandlung zu den erwarteten Wacker-Produkten 2 a und 3 a beobachtet. Der Wechsel des Lösungsmittels zu tBuOH führte zu einer geringen, aber nachweisbaren Menge an Carbonylprodukten (Eintrag 2). Während dieses Ergebnis die prinzipielle Durchführbarkeit der Alkenoxidation zeigt, war die erforderliche Reoxidation des anfallenden Pd0 in diesem Fall nicht erfolgreich. Um diese Einschränkung zu beheben, wurde eine Reihe von Oxidationsmitteln wie Benzochinon (BQ) oder tert-Butylhydroperoxid getestet, allerdings ohne Verbesserung (Einträge 3 & 4). Bei Verwendung eines Palladiumnitritkomplexes anstelle von Palladiumchlorid,17 erwies sich die Reoxidation unter aeroben Bedingungen mit CuCl2 als Mediator dagegen als erfolgreich. So wurde das umgelagerte Keton 2 a in 77 % Ausbeute zusammen mit 12 % des entsprechenden Aldehyds 3 a erhalten, was die Bedeutung von Nitrit für diese Oxidation unterstreicht (vgl. Eintrag 5 zu Eintrag 2). Das Lösungsmittel erwies sich als ausschlaggebend für die Selektivität von Keton zu Aldehyd, wobei bessere Ergebnisse mit Alkoholen wie iPrOH oder EtOH erzielt wurden (Einträge 6 und 7). Ähnliche Beobachtungen wurden kürzlich von Kang und Mitarbeitern gemacht.7c Leider ging die verbesserte Keton-Selektivität auf Kosten einer insgesamt geringeren Ausbeute. Es wurde jedoch festgestellt, dass die Aktivität wiederhergestellt werden kann, indem das Oxidationsmittel auf tBuONO und der Katalysator wieder auf den handelsüblichen Palladiumdichlorid-Acetonitril-Komplex umgestellt wird, wodurch eine Ausbeute von 95 % von Cyclopentanon 2 a erzielt wurde. Die Bildung des Aldehyd-Nebenprodukts wurde währenddessen vollständig unterdrückt (Eintrag 8). Eine erfolgreiche Reoxidation von Pd0 durch Nitrit wurde bereits früher beschrieben.18 Weitere Untersuchungen der Reagenzien ergaben, dass tBuONO in diesem Prozess als terminales Oxidationsmittel fungiert (Eintrag 9), Wasser für einen effizienten Umsatz erforderlich ist (Eintrag 10) und Chlorid als Gegenion zum Palladiumkatalysator wesentlich ist (Eintrag 11). Schließlich konnte die Reaktionszeit auf 3 Stunden verkürzt und die Katalysatorladung auf 5 mol-% gesenkt werden, ohne dass es zu Ausbeuteeinbußen kam (Eintrag 12).
|
|||||
# |
Katalysator |
Oxidans (Äq) |
Lösemittel |
Yield [%] |
2 a:3a[a] |
|---|---|---|---|---|---|
1 |
PdCl2 |
CuCl2 (0.4) O2 |
DMF/H2O |
– |
– |
2 |
PdCl2(MeCN)2 |
CuCl2 (0.4)b |
tBuOH |
13 |
81 : 19 |
3 |
PdCl2(MeCN)2 |
BQ (1.0) |
tBuOH |
<1 |
– |
4 |
PdCl2(MeCN)2 |
tBuOOH (1.0) |
tBuOH |
11 |
|
5 |
Pd(NO2)Cl(MeCN)2 |
CuCl2 (0.4)[b] |
tBuOH |
88 |
88 : 12 |
6 |
Pd(NO2)Cl(MeCN)2 |
CuCl2 (0.4)[b] |
iPrOH |
44 |
91 : 9 |
7 |
Pd(NO2)Cl(MeCN)2 |
CuCl2 (0.4)[b] |
EtOH |
18 |
>99 : 1 |
8 |
PdCl2(MeCN)2 |
tBuONO (1.0) |
EtOH |
95 |
>99 : 1 |
9 |
PdCl2(MeCN)2 |
tBuONO (0.2) O2 |
EtOH |
19 |
>99 : 1 |
10[c] |
PdCl2(MeCN)2 |
tBuONO (1.0) |
EtOH |
36 |
>99 : 1 |
11 |
Pd(OAc)2 |
tBuONO (1.0) |
EtOH |
<1 |
– |
12[d] |
PdCl2(MeCN)2 |
tBuONO (1.0) |
EtOH |
95 |
>99 : 1 |
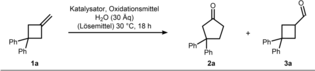
- Die Reaktionen wurden auf 0.1 mmol Maßstab in 1 mL Lösemittel [0.1 M] mit 10 mol % Katalysator durchgeführt [a] Die Ausbeute und Selektivität von 2 a und 3 a wurden mit 1H NMR des Reaktionsgemischs bestimmt; Mesitylen als interner Standard. [b] Atmosphärischer Sauerstoff wurde als terminales Oxidans verwendet. [c] Die Reaktion wurde wasserfrei unter Argonatmosphäre durchgeführt. [d] Reaktion wurde mit 5 mol % Katalysator durchgeführt und nach drei Stunden gestoppt.
Um einen Einblick in den Ursprung des Sauerstoffatoms von 1 a zu erhalten, wurde 18O-markiertes Wasser als massensensitiver Tracer verwendet (Schema 3a). Um eine Sauerstoffeinschleppung während der Aufarbeitung und Aufreinigung zu verhindern, wurde die Reaktion nach 2 Stunden durch Zugabe von Natriumborhydrid im Überschuss gestoppt. So wurde Cyclobutanol 4 a als einziges Produkt in 89 % Ausbeute isoliert. Massenspektrometrisch wurde ein 18O-Gehalt von 78 % nachgewiesen, was auf einen ersten Hydroxypalladierungsschritt hindeutet. Anschließend wurde das endständig 13C-markierte Methylencyclobutan 13C-1 a der Reaktionssequenz unterzogen, wobei Cyclopentanon 13C-2 a in Übereinstimmung mit einer 1,2-Kohlenstoffverschiebung erhalten wurde. Alle Versuche, ein Zwischenprodukt aus einer β-Kohlenstoff-Eliminierung abzufangen, wie z. B. die Durchführung der Reaktion unter einer CO-Atmosphäre, waren erfolglos. Dies deutet zwar auf eine konzertierte 1,2-Verschiebung hin, schließt aber einen schrittweisen Mechanismus wie in Schema 2 vorgeschlagen nicht aus.19

Markierungexperimente als Überprüfung der mechanistischen Hypothese.
Nachdem wir den Mechanismus und die kritischen Reaktionsparameter untersucht hatten, machten wir uns daran, die Anwendungsmöglichkeiten der Wacker Oxidation zu erkunden (Schema 4). 3-Phenyl-MCB (1 b) war ein brauchbares Substrat, wie die Bildung von 3-Phenyl-Cyclopentanon 2 b in 92 % Ausbeute zeigte. Sterische und elektronische Änderungen am Phenylring wurden unter den optimierten Bedingungen gut toleriert (2 c–2 h). Erwähnenswert ist die Bromsubstitution (2 h), die mit dem intermediär gebildeten Pd0 nicht interferierte. Heterozyklen wie Indol (2 i) waren ebenfalls mit dem oxidativen Reaktionsprotokoll kompatibel. Die Reaktion funktioniert ebenso effizient mit einer Reihe von 3,3-disubstituierten MCBs (2 j–2 m) und kann mit nur geringen Abweichungen im Grammmaßstab durchgeführt werden (2 j). Spirozyklische (2 n–2 o) und vollständig gesättigte Cyclopentanone (2 p) wurden durch die Wacker Oxidation in durchgehend ≥90 % Ausbeute erhalten. Interessanterweise wurde das 3-Cyano-Cyclopentanon 2 q in einer mäßigen Ausbeute von 34 % isoliert. Die niedrige Ausbeute kann durch die mögliche HCN-Eliminierung und die Flüchtigkeit der Produkte erklärt werden. Es ist wichtig zu beachten, dass in diesem einzigen Fall geringe Mengen (4 %) des entsprechenden Aldehyds durch NMR-Analyse des rohen Reaktionsgemischs nachgewiesen wurden. Im Gegensatz dazu wurden funktionelle Gruppen wie geschützte Amine, Ether, freie Alkohole und Ester ebenfalls untersucht, wobei die entsprechenden Cyclopentanone 2 r–2 w in guter bis ausgezeichneter Ausbeute ohne nachweisbare Aldehydbildung erhalten wurden. Es wurde ebenso festgestellt, dass die Reaktion auf Methylencyclobutane als Substrate beschränkt ist. Kleinere Ringe wie Methylencyclopropane sowie größere Ringe wie Methylencyclohexane durchliefen unter den optimierten Bedingungen keine Ringerweiterung. Die nicht umgesetzten Alkene wurden in beiden Fällen nachgewiesen (weitere Einzelheiten siehe Hintergrundinformationen).

Die Reaktionen wurden in EtOH [0.1 M] auf 0.3 mmol Maßstab mit 5 mol % Katalysator, 1 Äquiv. tBuONO, und 30 Äquiv. H2O durchgeführt. [a] Kleine Menge vom entsprechenden Aldehyd wurden bei diesem Substrat gefunden. [b] Die Reaktion wurde auf 0.18 mmol Maßstab durchgeführt. [c] Isoliert als Keton:Halbketal Gemisch (66 : 34 Verhältnis).
Nach der Erkundung des Substratspektrums interessierten wir uns für die Selektivität des Oxidationsprozesses. Es war nicht überraschend, dass die Reaktion hochselektiv war, wie die Oxidation von Dien 1 x zu Cyclopentanon 2 x in 91 % isolierter Ausbeute zeigte (Schema 5, oben). Darüber hinaus wurde 2-Benzyl-MCB 5 als mechanistische Probe verwendet, um die Migrationsneigung von sekundären gegenüber primären C-C-Bindungen zu testen. Es wurde eine geringfügige Bevorzugung des C-3-funktionalisierten Cyclopentanons 6 gegenüber seinem C-2-Gegenstück 7 beobachtet, was auf eine schnellere 1,2-Verschiebung der höher substituierten Bindung hindeutet (Schema 4b, Weg a). Prochirale MCBs wie 1 j haben das Potential für eine enantioselektive Desymmetrisierung und veranlassten uns, eine kleine Gruppe von Liganden zu testen (14 Liganden wurden getestet, siehe Hintergrundinformationen für weitere Details). Als Beweis für das Konzept wird der Pyridin-Oxazolin (Pyox)-Ligand L1 hervorgehoben, der das Cyclopentanon (+)-2 j in einem Enantiomerenverhältnis (er) von 63 : 37 liefert.20 Um die Reaktion zu einem angemessenen Umsatz zu bringen, musste die Temperatur auf 78 °C erhöht und das Chloridgegenion durch ein Perchlorat ersetzt werden. Dieses Ergebnis ist nicht nur die erste enantioselektive Desymmetrisierung eines MCB, sondern verleiht auch dem Schlüsselschritt, der durch eine Palladium-vermittelte Semipinakol-Umlagerung zustande kommt, weitere Glaubwürdigkeit.

Selektivitätsanalyse der Wacker Oxidation mit a) Alken anhängigen, b) α-substituierten und c) prochiralen MCBs. Ausbeute des Reaktionsgemischs ist in Klammern angegeben und wurde mittels 1H NMR mit Mesitylen als internen Standard bestimmt. [a] Regioisomerenverhältnis wurde aus Reaktionsgemisch mittels 1H NMR Spektroskopie bestimmt. [b] Enantiomerenverhältnis wurde mittels HPLC unter Verwendung einer chiralen Säule bestimmt.
Zusammenfassung
Zusammenfassend lässt sich feststellen, dass die ketonselektive Wacker Oxidation von 1,1-disubstituierten Alkenen über eine semipinakolartige Umlagerung mit gespannten MCBs entwickelt werden konnte. Während der Optimierung stellte sich heraus, dass tert-Butylnitrit eine entscheidende Rolle bei der Aktivierung des Palladiumkatalysators durch die Bereitstellung von Nitrit und bei der Vermittlung einer effizienten Reoxidation von Pd0 spielt. Auf diese Weise kann eine Reihe von Cyclopentanonen mit guter Toleranz gegenüber funktionellen Gruppen bei 30 °C in nur 3 Stunden zugänglich gemacht werden. Das stereochemische Ergebnis der Reaktion wurde daraufhin untersucht und ein geeigneter Pyox-Ligand identifiziert, der die Desymmetrisierung von prochiralen MCBs ermöglicht. Somit wird die bislang unerreichte enantioselektive Wacker-Oxidation realisiert, was einen neuen Zugang zu Cyclopentanonen als nützlichen chiralen Baustein eröffnet.
Danksagung
Wir bedanken uns für die großzügige Unterstützung der Johannes Gutenberg-Universität Mainz, der Westfälischen Wilhelms-Universität Münster und des Fonds der Chemischen Industrie (Liebig-Doktoranden-Stipendium an M.T. und Liebig-Stipendium an J.M.W.). Open Access Veröffentlichung ermöglicht und organisiert durch Projekt DEAL.
Interessenkonflikt
Die Autoren erklären, dass keine Interessenkonflikte vorliegen.
Open Research
Data Availability Statement
Die Daten, die die Ergebnisse dieser Studie unterstützen, sind in den Hintergrundinformationen zu diesem Artikel verfügbar.

